UNIT 13 PRINCIPLES OF GENE TECHNOLOGY
UNIT 13: PRINCIPLES OF GENE TECHNOLOGY
Key Unit Competence
Explain the principles of gene technology.
Learning ObjectivesBy the end of this unit, I should be able to:
–Define the term recombinant DNA.
–Explain that genetic engineering involves the extraction of genes from one organism
or the synthesis of genes, in order to place them in another organism(of the same or another species) such that the receiving organism expresses the gene product.
–Describe the properties of plasmids that allow them to be used in gene cloning.
–Explain the use of genes in fluorescent or easily stained substances as markers in gene technology.
–Describe the principles of the Polymerase Chain Reaction (PCR) to clone and amplify DNA(the role of Taq polymerase should be emphasized).
–Describe and explain how gel electrophoresis is used to analyse proteins and nucleic
acids, and to distinguish between the alleles of a gene (limited to the separation of polypeptidesand the separation of DNA fragments cut with restriction endonucleases).
–Explain the roles of restriction endonucleases, reverse transcriptase and ligasesin genetic engineering.
–Explain and outline, how microarrays are used in the analysis of genomes and indetecting mRNA in studies of gene expression.
–Interpret illustrations of the isolation and transfer of genes using plasmids in transgenicorganisms (bacteria, plant or an animal).
–Sequence the processes involved in the extraction and transfer of genes fromone organism to another.
–Interpret charts of the Polymerase Chain Reaction (PCR).
–Relate the mechanism of DNA replication to PCR and the amount of DNAproduced in a given period of time.
–Appreciate that the easy transfer of some plastids from one species of bacteria toanother may carry genes for antibiotic resistance.
–Acknowledge that advances in genetic engineering have enabled manipulation of genesto our advantage.
Introductory activity
Observe the figures below and respond to the questions that follow:
1. State and explain briefly the Chargaff’s rule of bases pairing based on the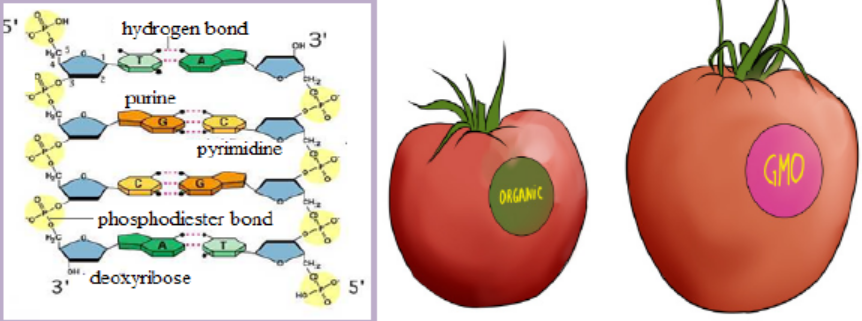
DNA structure shown above.
2. Describe briefly the gene expression starting by the DNA structure shown
above.
3. Summarize the main action done to transform organic tomato intogenetically modified organism (GMO), also called transgenic organism
13.1 Recombinant DNA and enzymes involved in genetic engineering
Activity 13.1
Using textbooks and or internet to answer the following questions.
1. Explain briefly the terms below:
a. Recombinant DNA
b. Transgenic organism
c. Enzyme2. Describe briefly the role of enzymes involved in genetic engineering
13.1.1 Recombinant DNA
A recombinant deoxyribonucleic acid (r DNA) is the DNA that contains genes from
more than one source. Examples of molecules produced from recombinant DNA
and that are important to humans include some pharmaceuticals like human insulin
and antibiotics.
Genetic engineering, also known as recombinant DNA technology or gene cloningor gene technology is the alteration of the genes in a living organism to produce a
genetically modified organism (GMO) with a new genotype. Various kinds of genetic
modification are possible and include:
– Inserting a foreign gene from one species into another in order to form a
transgenic organism,
– Altering an existing gene so that its product is changed and changing geneexpression so that it is translated more often or not at all.
13.1.2 Role of some enzymes in genetic engineering
The enzymes involved in gene manipulation include; restriction endonucleases(restriction enzymes), methylase, ligase and reverse transcriptase.
Restriction endonucleases
Different restriction enzymes, also called restriction endonucleases, exist and cut
the DNA molecule into fragments; their examples are shown in the table 13.1below.Table 13.1: List of some restriction enzymes and their respective recognition sites
Restriction enzymes are named according to the bacteria from which they originate.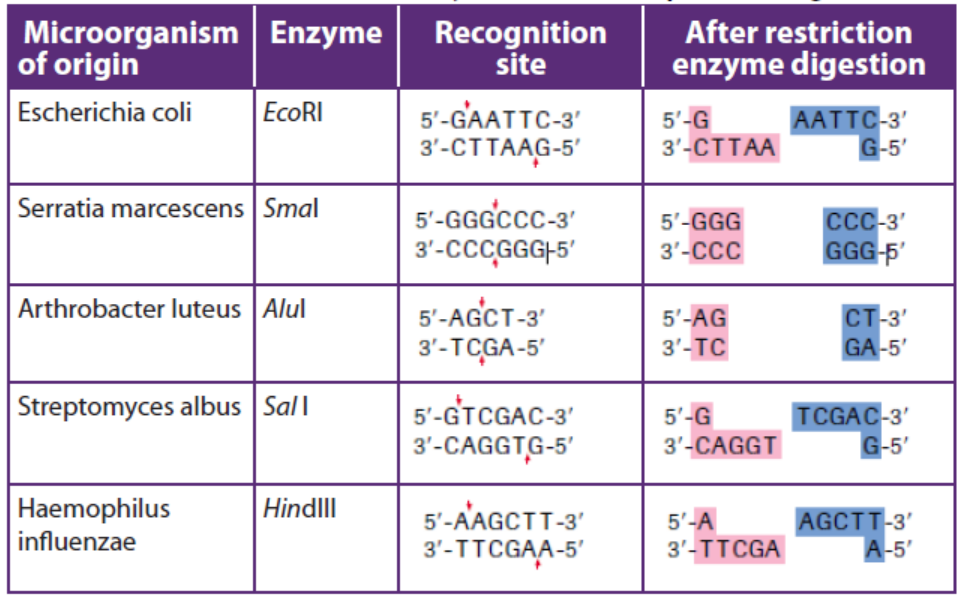
For example, the restriction enzyme BamHI is named as follows:
– B represents the genus Bacillus
– am represents the species amyloliquefaciens
– H represents the strain– I mean that it was the first endonuclease isolated from this strain
A commonly used tool in molecular biology is restriction endonucleases which
are molecular scissors that can cut double-stranded DNA at a specific base-pair
sequence. Each type of restriction enzyme recognizes a characteristic sequence of
nucleotides that is known as its recognition site. Most recognition sites are four to
eight base pairs long and are usually characterized by a complementary palindromicsequence.
For example, the restriction enzyme EcoRI binds to the following base-pair sequence:
5’-GAATTC-3’/3’-CTTAAG-5’. It is palindromic because both strands have the same
base sequence when read in the 5’ to 3’ direction. EcoRI scans a DNA molecule and
only stops when it is able to bind to its recognition site. Once bound, it disrupts,
via a hydrolysis reaction, the phosphodiester bond between the guanine and
adenine nucleotides on each strand. A phosphodiester bond is a covalent bond
located between a two sugar groups and a phosphate group; such bonds form the
sugar-phosphate backbone of DNA and RNA. Subsequently, the hydrogen bonds of
complementary base pairs between the cuts are disrupted. The result is a cut withina DNA strand, producing two DNA fragments where once there was only one.
So, in cleavage of DNA sequence using restriction enzyme EcoRI:EcoRI scans the DNA molecule:
The ends of DNA fragments produced from a cut by different restriction endonucleases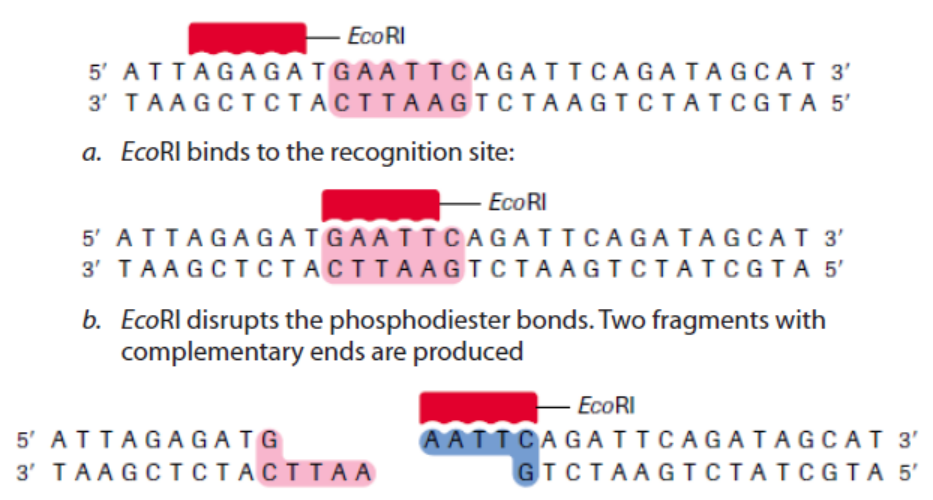
differ, depending on where the phosphodiester bonds are broken in the recognition
site. In the example in Table 13.1, EcoRI produces sticky ends; that is, both fragmentshave DNA nucleotides that are now lacking their respective complementary bases.
These overhangs are produced because EcoRI cleaves between the guanine and
the adenine nucleotide on each strand. Since A and G are at opposite ends of the
recognition site on each of the complementary strands, the result is the overhang.
In few words, sticky ends are fragment end of a DNA molecule with short singlestranded overhangs, resulting from cleavage by a restriction enzyme.
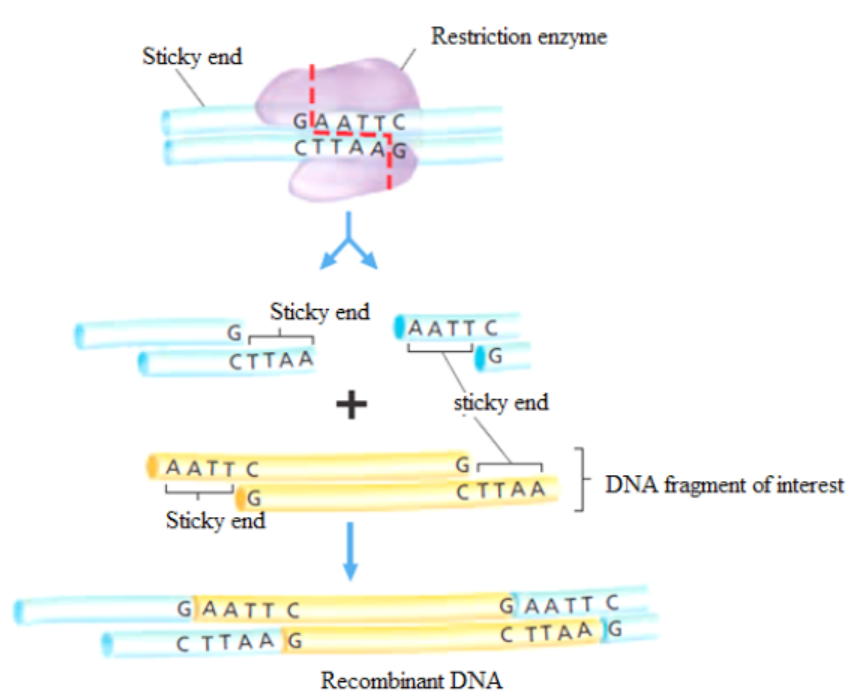
Figure 13.2: Cutting DNA by restriction enzymes and rDNA formation.
b. Methylases
These are enzymes that add a methyl group (CH3) to one of the nucleotides found in
a restriction endonuclease recognition site, altering its chemical composition. They
allow the molecular biologist to protect a gene fragment from being cleaved in anundesired location.
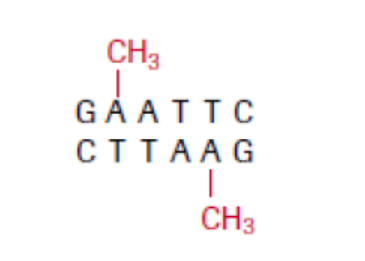
Figure 13.3: At a methylated EcoRI site, EcoRI restriction enzyme is no longer able to cut.
c. DNA ligase
This enzyme repairs broken DNA by joining two nucleotides in a DNA strand. It is
commonly used in genetic engineering to do the reverse of a restriction enzyme that
is to join together complementary restriction fragments. The sticky ends allow two
complementary restriction fragments to harden, but only by weak hydrogen bonds,
which can quite easily be broken by gentle heating. The backbone is still incomplete.
DNA ligase completes the DNA backbone by forming covalent bonds. T4 DNA ligase
is an enzyme that originated from the T4 bacteriophage and which is used to join
together DNA blunt or sticky ends. So, DNA ligase is able to join complementarysticky ends produced by the same restriction enzyme via a condensation reaction:
a. Complementary sticky ends produced by HindIII.
b. Hydrogen bonds form between complementary bases. DNA ligase
reconstitutes the phosphodiester bond in DNA backbones.
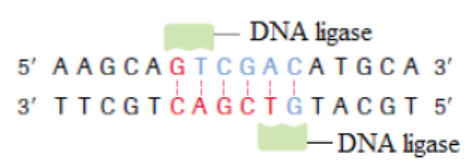
c. If fragments are not complementary, then hydrogen bonds will not form.
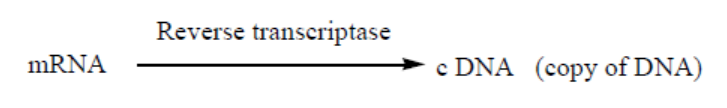
d. Reverse transcriptase
Reverse transcription is a process whereby a mRNA is converted into c DNA
(complementary DNA, also called copy of DNA). It requires the enzymes calledreverse transcriptase. It is shown by this reaction:
Application 13.1
1. Write the following abbreviations in full: DNA, GMO and RNA.
2. Explain the nomenclature of the enzyme EcoRI.
3. Distinguish between sticky ends and blunt ends.4. Discuss the role of T4 DNA ligase.
13.2 Properties of plasmids and gene manipulation
Activity 13.2
Use different biology textbooks or internet to respond to the following
questions.
1. Identify any 3 properties of plasmids.
2. Explain the role of vectors in genetic engineering.3. Elaborate the main steps of gene manipulation
13.2.1. Properties of plasmids
A plasmid is a genetic structure, in some cells, that can replicate independently
of the chromosomes; it is typically a small circular DNA strand in the cytoplasm
of a bacterium or protozoan. Plasmids are much used in the laboratory duringmanipulation of genes.
Figure 13.4. The structure of the Tumor – inducing plasmid (Ti plasmid) of Agrobacterium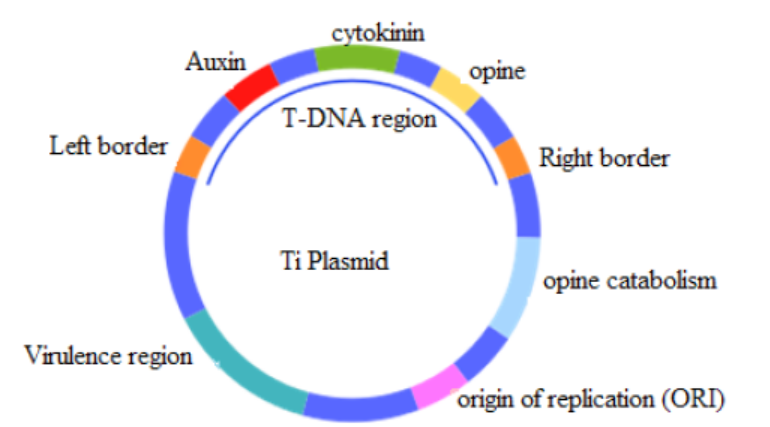
tumefaciens and Agrobacterium rhizogenes
The properties of plasmids are:
– It is big enough to hold the desired gene.
– It is circular (or more accurately a closed loop), so that it is less likely to be
broken down.
– It contains control sequences, such as a transcription promoter, so that the
gene will be replicated or expressed.
– It contains marker genes, so that cells containing the vector can be identified.
Plasmids are not the only type of vector that can be used. Viruses can also be used
as vectors. Another group of vectors are liposomes, which are tiny spheres of lipidcontaining the DNA.
13.2.2. Gene manipulation
Genetic manipulation is a process done to use the genome of an organism in order
to produce desired traits. A genome is the complete set of genes or genetic material
present in an organism.
Genes are pieces of DNA, that carry genetic information which determines all the
characteristics of an individual such as eye colour, size, ability to resist disease, etc.
Each gene contains the information required to build specific proteins needed in an
organism.
The human genome contains 20,687 protein-coding genes.
The overview of gene transfer, resulting in genetically modified organisms (GMO)
also called transgenic organisms such as bacteria or animals or plants havingforeign gene inserted into them, is shown below:
1. Generation of DNA fragments using restriction endonucleases:
– Appropriate restriction endonucleases need to be used to ensure that the
gene fragment in question is excised completely from the source DNA.
– More than one restriction endonuclease may be used at one time.
2. Construction of a recombinant DNA molecule:
– The target gene fragment is ligated to a DNA vector (plasmids are one example)
and is now recombinant DNA.
– The vector can replicate autonomously in an appropriate host organism.
3. Introduction into a host cell:
– Bacterial host cells can be manipulated to take up the recombinant DNA usingelectroporators, gene guns or classical transformation protocols.
– Once the bacterium takes up the recombinant DNA, it is referred to as being
transformed.
4. Selection:
– Cells that have been successfully transformed with the recombinant DNA must
be isolated.
– The desired cells are usually chemically selected by the presence of a marker
(e.g. antibiotic resistance) on the vector.
– Growth of colonies on media containing the chemical indicates successful
transformation of the recombinant DNA vector.
– Individual colonies are isolated from media containing the chemical and are
grown in culture to produce multiple copies (clones) of the incorporated
recombinant DNA. Different gene manipulations are illustrated under the
heading 13.3.
To perform these gene manipulation steps, the genetic engineer needs a tool kitconsisting of:
1. Enzymes, such as restriction endonucleases (restriction enzymes), ligase and
reverse transcriptase
2. Vectors, including plasmids and viruses3. Genes coding for easily identifiable substances that can be used as markers.
Application 13.2
1. Identify the vectors that are used in genetic engineering.
2. Find the components of a genetic engineering tool kit.
3. Differentiate between a gene and a genome.4. Explain the second step of gene manipulation.
13.3 Transfer of genes using plasmids in transgenic organisms
Activity 13.3
Using different Biology textbooks or internet:
1. What is meant by a pathogenic bacterium
2. Explain the role of a gene marker in genetic engineering.
3. Distinguish between bacterial transduction and transformation
4. Explain briefly the steps of formation of a transgenic plant
5. Draw and interpret the chart of about the transfer of DNA from
eukaryotic cell to a bacterial cell using a plasmid.
6. By diagrams, show how a transgenic organism such as a transgenicplant and a clone are produced.
The production of genetically modified organisms (GMO), also called transgenic
organisms, is a multistage process which can be generally summarized as follows:
– Identification of the gene of interest.
– Isolation of the gene of interest.
– Cutting of gene of interest and opening of plasmid with restriction enzymes in
order to have sticky ends
– Associating the gene with an appropriate promoter and poly -A sequence and
insertion into plasmids.
– Multiplying the plasmid in bacteria and recovering the cloned construct for
injection.
– Transference of the construct into the recipient tissue, usually fertilized eggs.
– Integration of gene into recipient genome.
– Expression of gene in recipient genome.– Inheritance of gene through further generations.
13.3.1. Extraction, purification, isolation and transfer of genes usingplasmids into bacteria
The normal gene coding for a particular protein is extracted from an organism; it
is isolated and transferred into a plasmid of a bacterium. This plasmid becomes a
recombinant DNA that is introduced into that bacterium. This bacterium becomes
a transgenic bacterium. An example of the sequence of the processes involved
in the extraction and transfer of genes from one organism to another is illustratedbelow.
Process 1: Extraction and purification of DNA containing an interest gene
is required for a variety of molecular biology applications. Its process issummarized below.
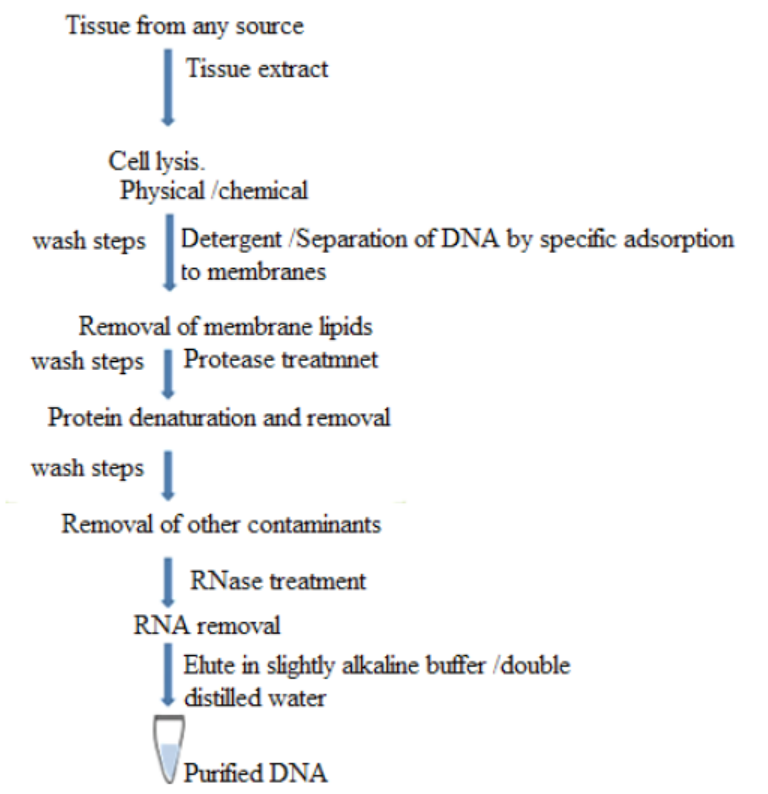
Figure 13.5: Basic steps involved in all DNA extraction methods
The purification of DNA from cell extract occurs in this way:
– The standard way to deproteinize a cell is to add phenol or a 1:1 mixture of
phenol and chloroform.
– The organic solvents precipitate proteins but leave the nucleic acids (DNA and
RNA) in an aqueous solution.
– The result is that is the cell extract is mixed gently with the solvent, and the
layers then separated by centrifugation, precipitated protein molecules are
left as a white coagulated mass at the interface between the aqueous and
organic layers.
– The aqueous solution of nucleic acids can then be removed with a white
pipette.
– Cell extract is treated with protease such as pronase or proteinase K before
extraction.
– These enzymes will break polypeptides into smaller units thus making phenol
easier to remove them.
– The only effective way to get rid of RNA is the use of ribonuclease enzyme
which will rapidly degrade the molecules into ribonucleotide subunits. As
DNA is purified, also its genes are purified.
The Concentration of DNA samples is carried out in this way:
– The most frequently used method of concentration is ethanol precipitation.
– In the presence of salt and a temperature of -20 oC or less absolute ethanol
with efficiently precipitate polymeric nucleic acids.
– With 2 thick solution of DNA, the ethanol can be layered on the top of the
sample.
– A spectacular trick is to push a glass rod through the ethanol into the DNA
solution.
– When the rod is removed, DNA molecules will adhere and be pulled out of thesolution in the form of long fiber.
Figure 13.6. Practical summary of DNA extraction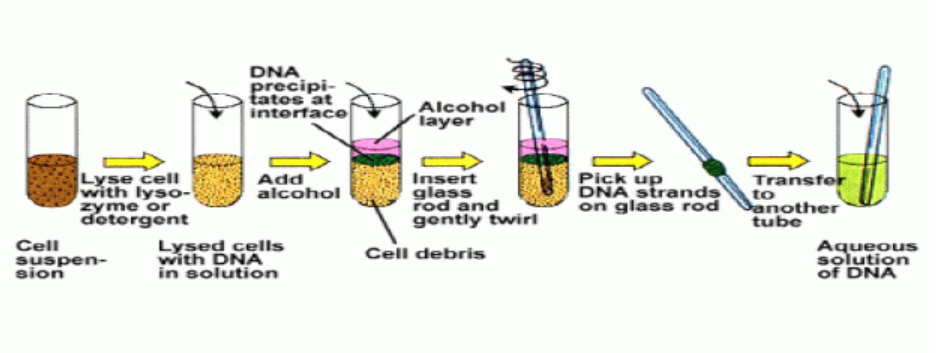
After getting DNA, it is possible to remove the gene from it, by a restriction enzyme, in
order to use it for a particular purpose. For example, normal insulin gene is removedfrom human cell as shown in the figure below.
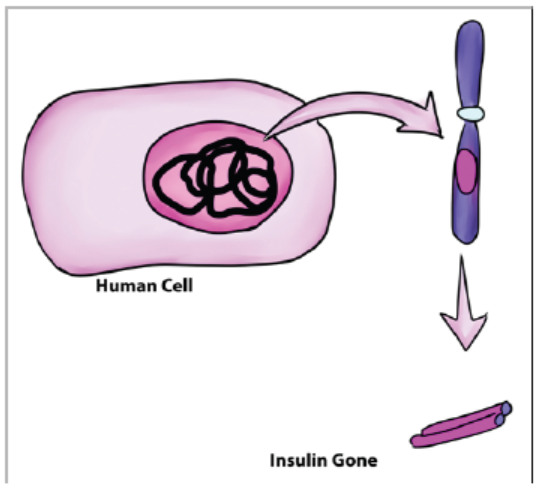
Figure 13.7: Removal of insulin gene from human cell
Process 2: Summary of transfer of insulin gene using plasmids into bacteria
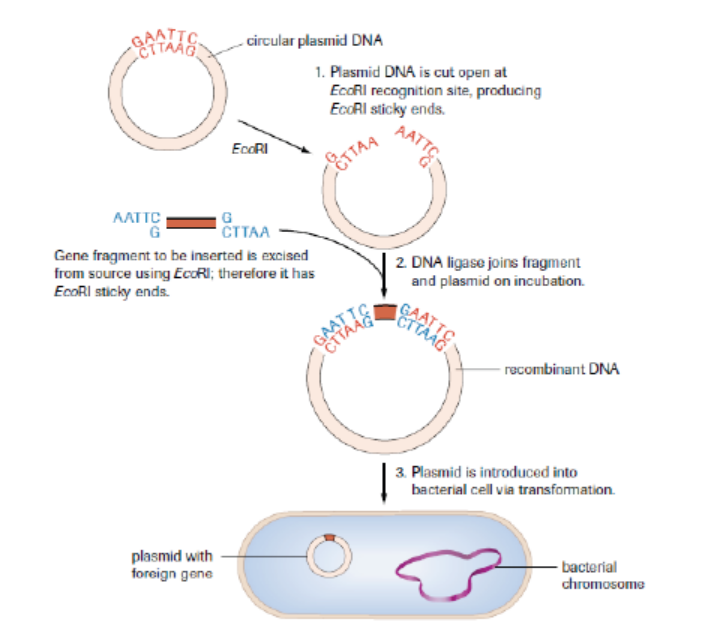
Figure 13.8: A foreign gene is introduced into a plasmid of a bacterium to form a recombinant DNA
The plasmid is now an example of recombinant DNA, which can be introduced into
a bacterial cell to produce numerous copies (clones) of the gene. As the inserted
gene codes for insulin, a hormone that reduces the blood glucose level, and this
gene functions normally as expected, the product (insulin) may also be retrievedand used for therapeutic purposes in which it is given to diabetic people.
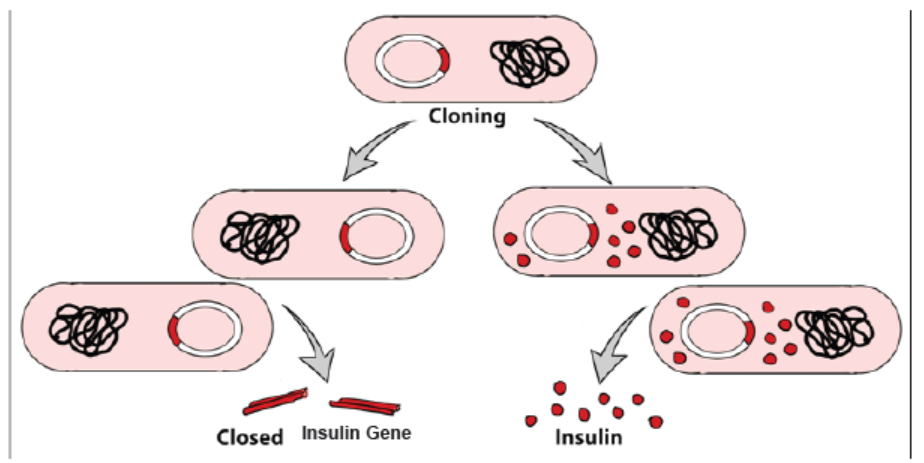
Figure 13.9: Gene cloning after a bacteriumpr otadkuecst iuopn a recombinant DNA (plasmid) and insulin
In nuclear biology and molecular biology, a marker gene is a gene used to determineif a nucleic acid sequence has been successfully inserted into an organism›s DNA.
13.3.2 Use of Agrobacterium tumefaciens to transfer genes in plants
Agrobacterium is a bacterium that uses a horizontal gene transfer (HGT). HGT is
the transfer of DNA between different genomes. HGT can occur in bacteria through
transformation, conjugation and transduction. However, it is also possible for HGT
to occur between eukaryotes and bacteria. Bacteria have three ways of transferringbacteria DNA between cells:
1. Transformation: The uptake and incorporation of external DNA into the
cell thereby resulting in the alteration of the genome.
2. Conjugation: The exchange of genetic material through cell-to-cell contact
of two bacterial cells. A strand of plasmid DNA is transferred to the recipient
cell and the donor cell then synthesis DNA to replace the strand that was
transferred to the recipient cell.
3. Transduction: A segment of bacterial DNA is carried from one bacterial
cell to another by a bacteriophage. The bacteriophage infects a bacterial
cell and takes up bacterial DNA. When this phage infects another cell, it
transfers the bacterial DNA to the new cell. The bacteria can then become a
part of the new host cell.
Agrobacterium also has the ability to transfer DNA between itself and plants
and is therefore commonly used in genetic engineering. The process of usingAgrobacterium for genetic engineering is illustrated in the diagram below.
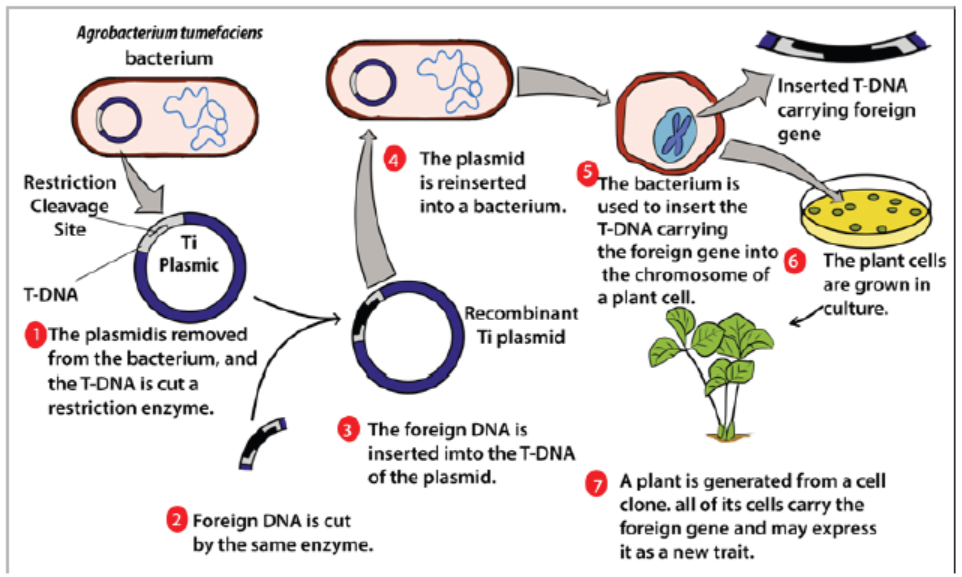
Figure 13.10: Process of formation of a transgenic plant
Summary of formation of a transgenic plant:
– The agrobacterium cell contains a bacterial chromosome and a Tumor inducing
plasmid (Ti Plasmid).
– The Ti plasmid is removed from the agrobacterium cell and a restriction
enzyme cleaves the T-DNA restriction site. The transfer DNA (T-DNA) is the
transferred DNA of the tumor-inducing plasmid of some species of bacteria
such as Agrobacterium tumefaciens
– The T-DNA is transferred from bacterium into the host plant›s
nuclear DNA genome.
– Next foreign DNA, which is also cleaved by the same enzyme, is inserted into
the T -DNA at the site that was cleavage site.
– The modified plasmid is then reinserted in the agrobacterium and the
bacterium inserts the T-DNA, which now carries a foreign gene into the plant
cell.
– The plant cell is then cultured and results in a new plant that has the foreignDNA trait.
13.3.3 Transfer of genes into animals
In reproductive cloning, researchers remove a mature somatic cell, such as a skin
cell, from an animal that they wish to copy. They then transfer the DNA of the donoranimal’s somatic cell into an egg cell, or oocyte, that has had its own DNA-containing
nucleus removed. For example, the cell used as the donor for the cloning of Dolly
sheep was taken from a mammary gland and the production of a healthy clone
therefore proved that a cell taken from a specific part of the body could recreate awhole individual.
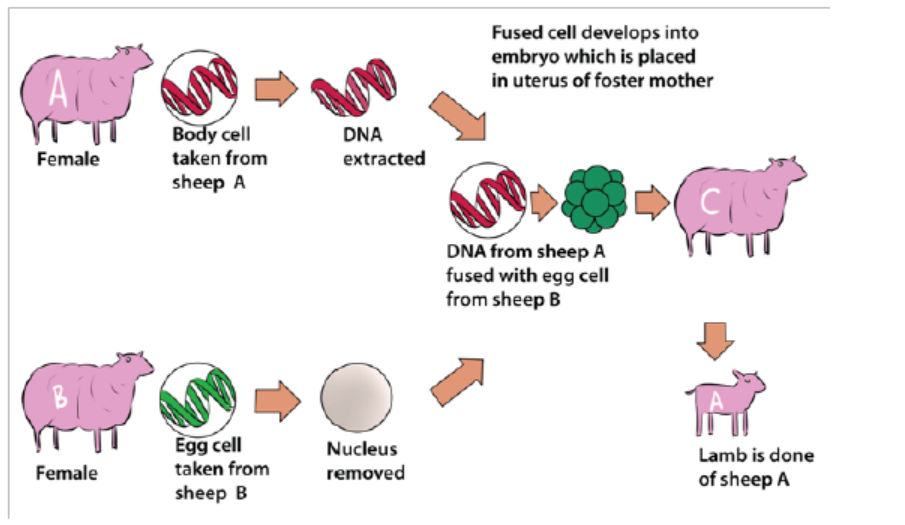
Figure 13.11: The cloning process that produced transgenic Dolly sheep
13.3.4 Transformation of harmless bacteria to pathogenic bacteria andresistant bacteria
A pathogenic bacterium is a bacterium which is capable of causing a disease.
An example of harmful or pathogenic bacterium is Vibrio cholerae which causes
cholera. A harmless bacterium can become pathogenic bacterium due to certain
factors. The discovery of DNA and the genetic code led scientists to determine that
some bacteria were resistant to particular antibiotics because of inserted genes that
rendered bacteria unaffected by the effects of some antibiotics. This gene insertion
can be done naturally between bacteria or artificially by biotechnologists.
Antibiotic resistance, also known as drug resistance, is the ability of bacteria
and other microorganisms to resist the effects of an antibiotic to which they were
once sensitive. Since bacteria are ubiquitous in the colon, conjugation is constantly
occurring. This conclusion has been supported by bacteria in different genera
containing homologous DNA plasmids. Therefore, horizontal gene transfer can occur
between different species or within a population. This can become problematic if
harmful bacteria that have been artificially selected for antibiotic resistance happen
to be in the colon, where bacteria can transfer the resistance gene to other species
of bacteria. Typically, this is not a problem because most bacteria are not harmful,
unless bacteria that are a public health concern happen to receive a resistance gene.
Individuals that have previously taken antibiotics are less responsive to treatmentbecause their bodies contain more antibiotic resistant bacteria.
These bacteria received these genes from disease - causing microbes that transferred
a resistance gene through conjugation or transformation. The harmless bacteria
that are resistant to antibiotics can then pass this gene to harmful bacteria that do
not yet have antibiotic resistance. Thus, horizontal gene transfer allows bacteria
to indirectly become resistant to antibiotics. Transformation and conjugation
contribute to increasing frequencies of antibiotic resistant genes because of genes
transferring between different species. The gene transfer can transform harmlessbacteria into pathogenic bacteria which can cause diseases.
The prevention of antibiotic-resistant infections includes:
– Do not take antibiotics for viral infections.
– Complete your prescribed course of treatment exactly as instructed by your
healthcare provider. Do not stop taking your medicine even if you feel better,
and do not save any antibiotics for future use.
– Do not take someone else’s antibiotics because different kinds of antibioticstreat different types of bacterial infections.
Application 13.3
1. Identify any bacterium involved in formation of transgenic plant.
2. Explain briefly the cloning of a sheep.
3. Describe briefly one cause of antibiotic resistance.
4. Discuss how biotechnologists might transform harmless bacteria topathogenic forms in the course of their studies.
13.4 Non-biological methods of gene transfer
Activity 13.4
Search from Biology textbooks and internets and answer to the following
questions:
1. Identify the non- biological methods of gene transfer
2. Explain the procedure of carrying out biolistics
3. Describe the disadvantages of vacuum infiltration.
Different non-biological methods, also called physical methods or direct methods
of gene transfer exist and include genetic transformation, shock wave-mediated
genetic transformation, electroporation, biolistic, vacuum infiltration, silicon carbidewhisker and laser microbeams.
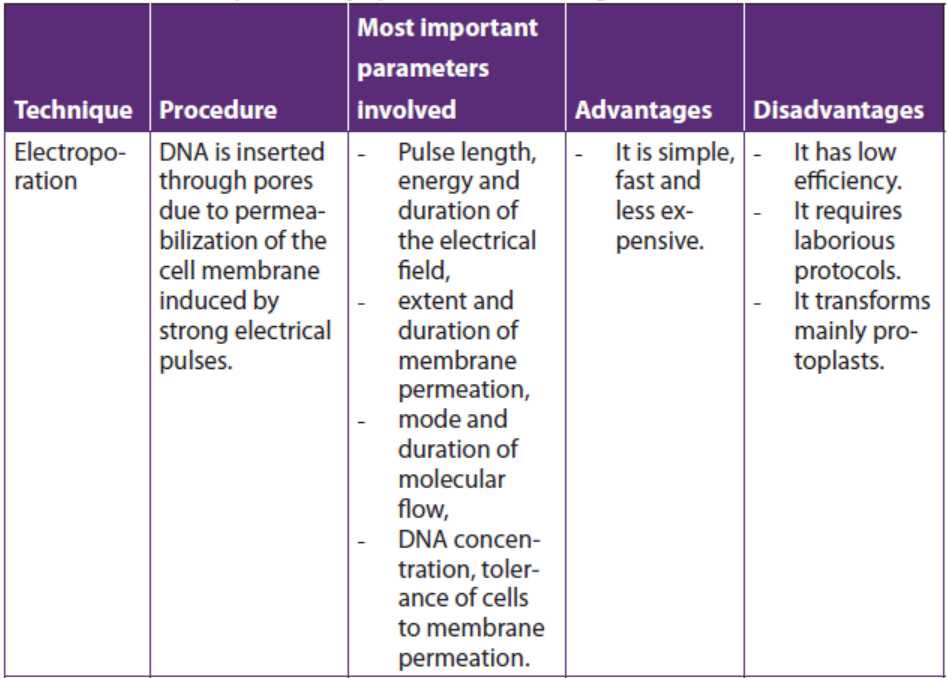
a. Electroporation
Electroporation is a method of transformation via direct gene transfer. In this
technique, a mixture containing cells and DNA is exposed to very high voltage
electrical pulses (4000 – 8000 Volts/cm) for very brief time periods (few milliseconds).
It results in formation of transient pores in the plasma membrane, through whichDNA seems to enter inside the cell and then nucleus.
Figure 13.12: Electroporation: (a) Diagram showing formation of transient pores in cell membrane on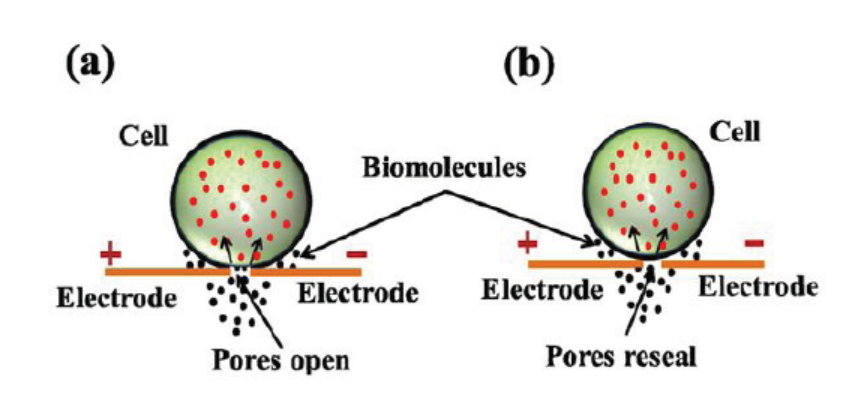
applying electrical pulse, (b) Entry of DNA inside the cell and sealing of pores afterwards.
A suspension of cells with plasmid DNA is taken in an electroporation cuvette
placed between electrodes and electrical pulses are applied. Temporary micropores
are formed in cell membranes which allow cells to take up plasmid DNA leading tostable or transient DNA expression.
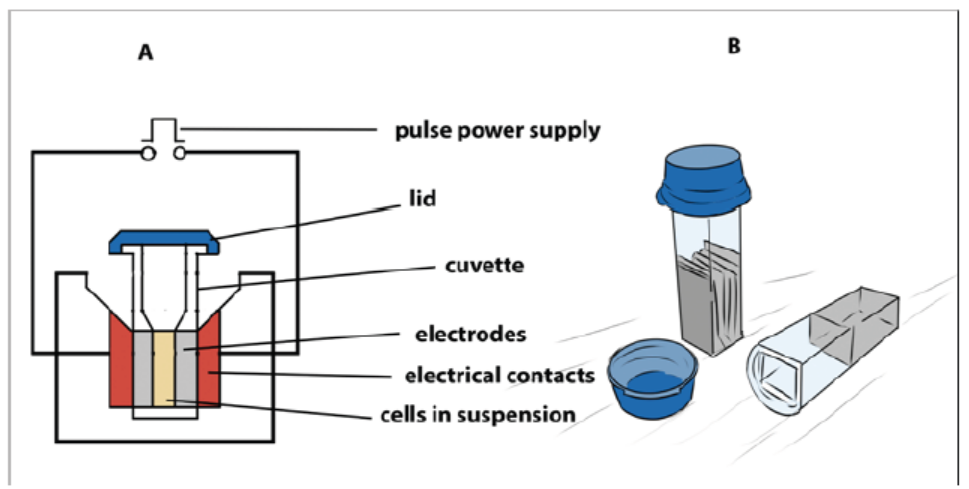
Figure 13.13: (A) Main components of an electroporator (B) Cuvettes used for electroporation
Cells which are arrested at metaphase stage of cell cycle are especially suitable for
electroporation as these cells have absence of nuclear envelope and an unusual
permeability of the plasma membrane. Protoplasts are used for electroporation of
plant cells as thick plant cell walls restrict movement of DNA. The electroporation
method was originally developed for protoplasts, but has given equally good results
with cells and even tissues with easy recovery of regenerated plantlets. Immature
zygotic embryos and embryogenic cells have also been used for electroporation to
produce transgenic maize.
Transformation of protoplast is associated with low transient expression
of transgenes as compared to organized tissues and low regeneration frequency
especially in monocotyledonous plants. The electrical field and chemical substances
applied to disorganize cell walls reduce the viability and capability of division of
protoplasts.
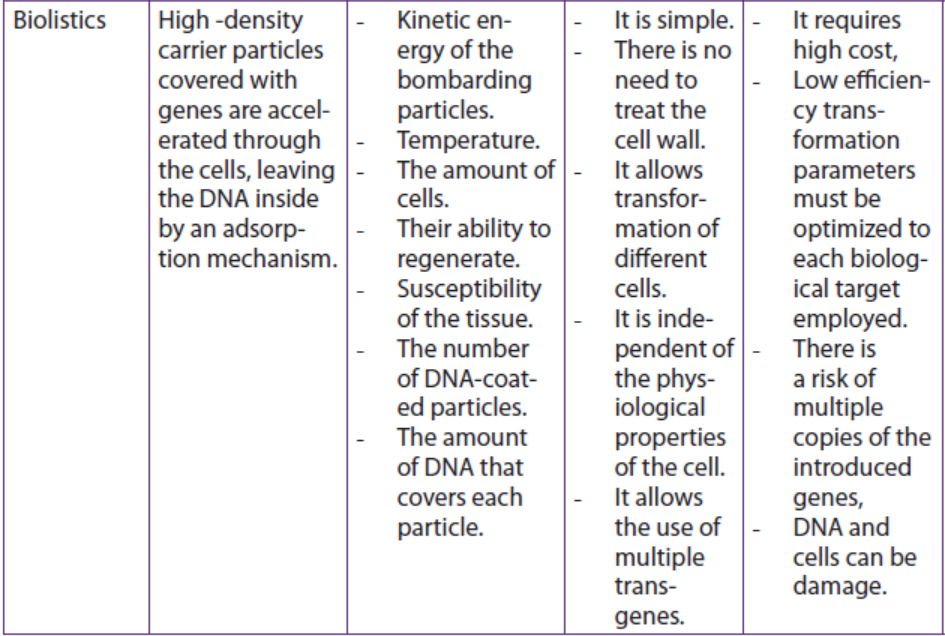
b. Biolistics
Biolistics, also called micro projectile or particle bombardment, is a method wherecells are physically impregnated with nucleic acids or other biological molecules.
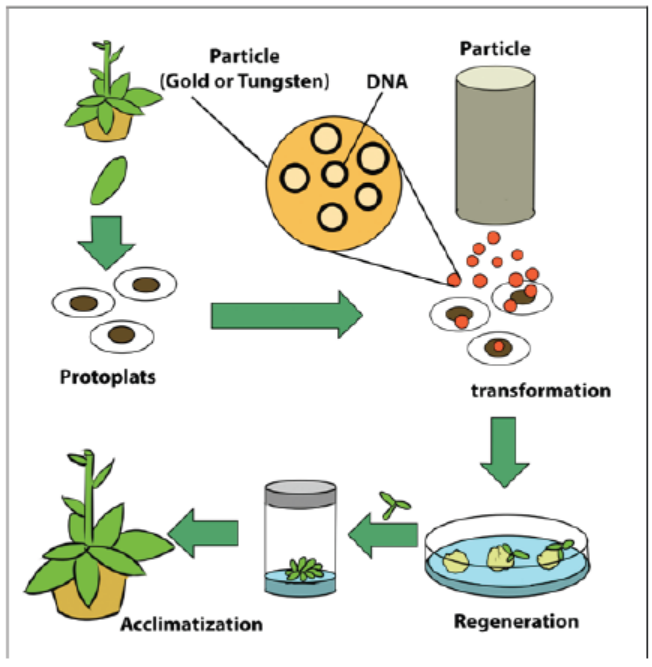
Figure 13.14: Particle bombardment method of plant transformation
The main steps of a biolistic method are:
– Isolation of protoplasts.
– Injection of DNA-coated particles using particle gun.
– Regeneration of transformed protoplasts into plantlets.
– Acclimatization of regenerated plantlets in a greenhouse.
A biolistic particle delivery system is a device for plant transformation where cells
are bombarded with heavy metal particles coated with DNA/RNA. This technique
was invented by John Stanford in 1984 for introduction of DNA into cells by physical
means to avoid the host-range restrictions of Agrobacterium. Agrobacteriummediated
genetic transformation system works well for dicotyledonous plants
but has low efficiency for monocots. Biolistic particle delivery system provides an
effective and versatile way to transform almost all type of cells. It has been proven
to be a successful alternative for creating transgenic organisms in prokaryotes,mammalian and plant species.
c. Microinjection
The process of using a fine glass micropipette to manually inject transgene atmicroscopic or borderline macroscopic level is known as microinjection.
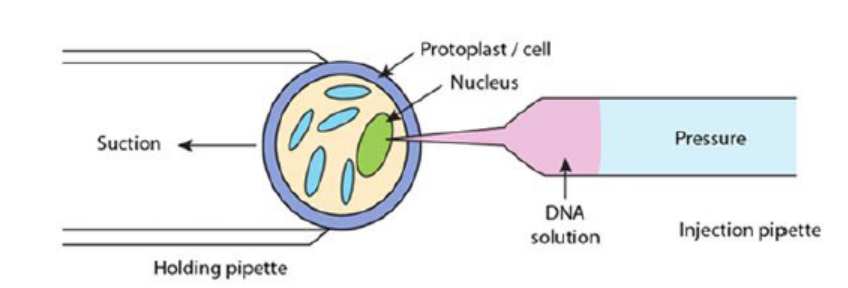
Figure 13.15: Illustration of microinjection method
The transgene, in the form of plasmids, cosmids, phage or PCR products, can be
circular or linear and does not need to be physically linked for injection. Microinjection
involves direct mechanical introduction of DNA into the nucleus or cytoplasm using
a glass microcapillary injection pipette. The protoplasts are immobilized in low
melting agar, while working under a microscope, using a holding pipette and suction
force. DNA is then directly injected into the cytoplasm or the nucleus. The injected
cells are then cultured in vitro and regenerated into plants. Successful examples of
this process have been shown in rapeseed, tobacco and various other plants.
Stable transformants can be achieved through this method but it requires technical
expertise and is a time consuming process. Also, microinjection has achieved only
limited success in plant transformation due to the thick cell walls of plants and a lack of
availability of a single-cell-to-plant regeneration system in most plant species. In this
technique, a traditional compound microscope (around 200x magnification) or an
inverted microscope (around 200x magnification) or a dissecting stereomicroscope
(around 40-50x) is used. The microscope target cell is positioned, cell membrane and
nuclear envelope are penetrated with the help of two micromanipulators. Onemicromanipulator holds the pipette and another holds the micro capillary needle.
The two types of microinjection systems are constant flow system and pulsed flow
system.
– In the constant flow system, the amount of sample injected is determined by
the duration for which needle remains in the cell. The constant flow system is
relatively simple and inexpensive but outdated.
– The pulsed flow system has greater control over the volume of substance
delivered, needle placement and movement and has better precision. This
technique results in less damage to the receiving cell; however, the componentsof this system are quite expensive.
d. Whiskers methods
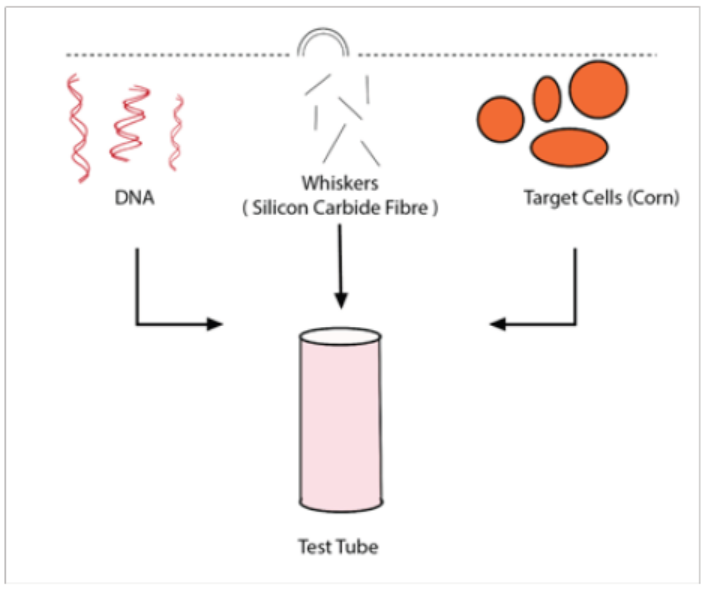
Figure 13.16: Whiskers methods
In this method, silicon carbide fibers are mixed in a vortex with a suspension of
tissue and DNA allowing introduction by abrasion. Maize (Zea mays) and tobacco
(Nicotiana tabacum) tissue cultures were transformed using silicon carbide fibers to
deliver DNA into suspension culture cells. DNA delivery was mediated by vortexingcells in the presence of silicon carbide fibers and plasmid DNA.
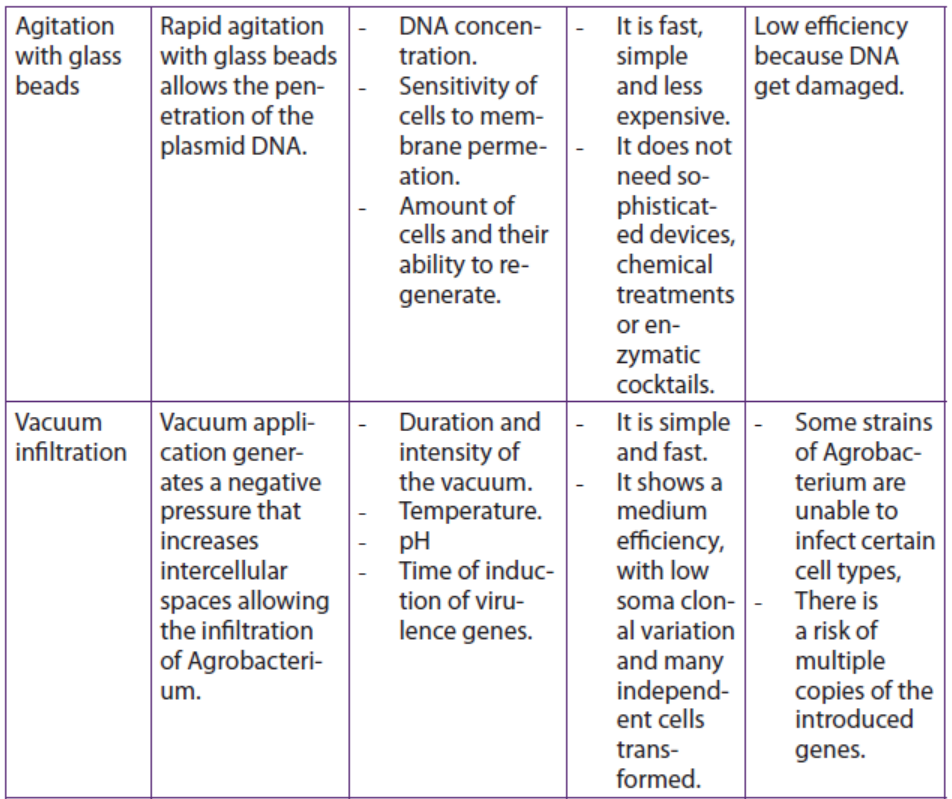
e. Vacuum infiltration method
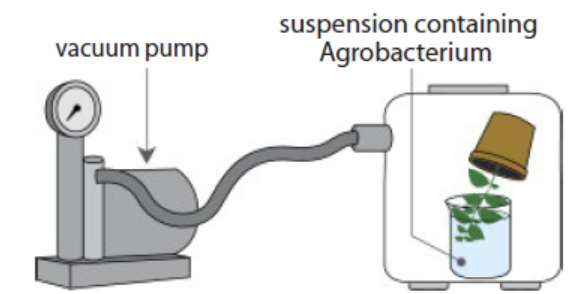
Figure 13.17: Vacuum infiltration methods
In this method, a vacuum pump generates a negative pressure that increasesintercellular spaces allowing the infiltration of Agrobacterium.
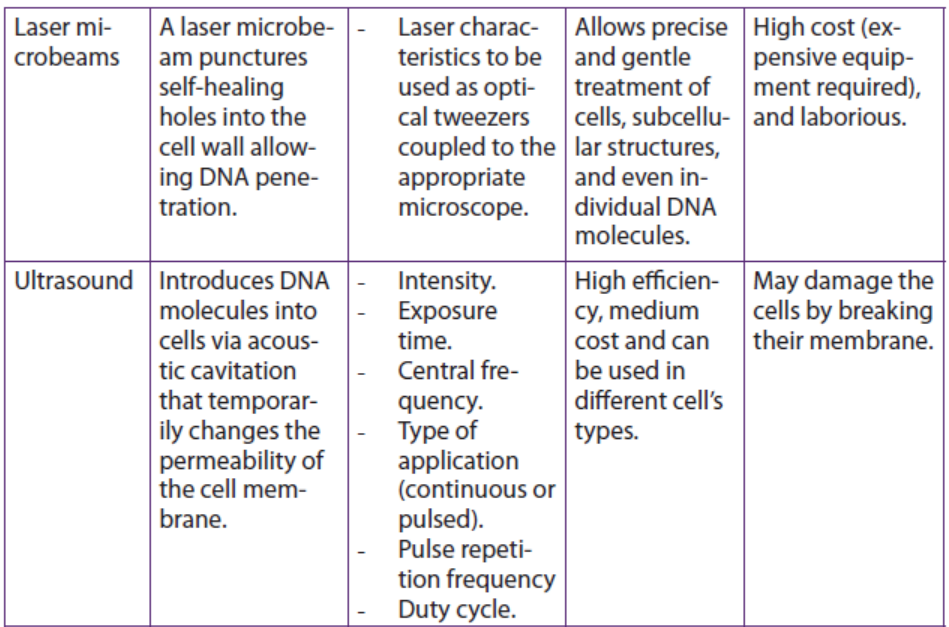
f. Laser microbeams method
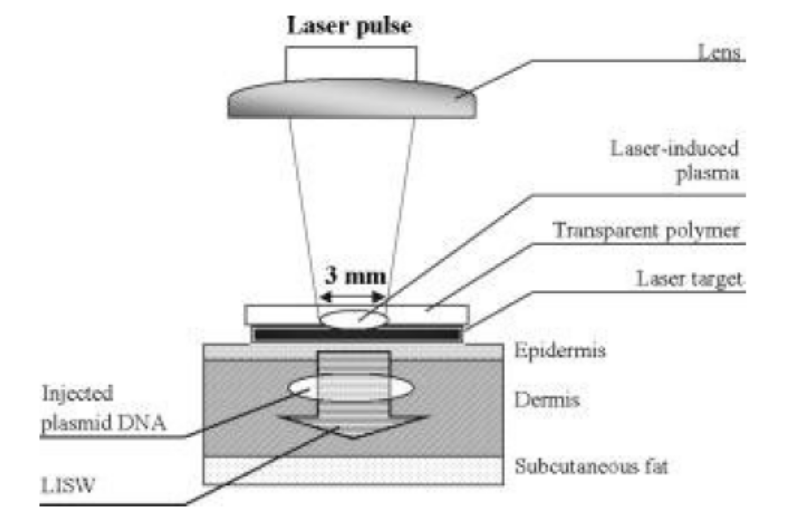
Figure 13.18. Laser microbeams method
In this method, a laser microbeam punctures self-healing holes into the cell wallallowing DNA penetration.
g. Ultrasound method
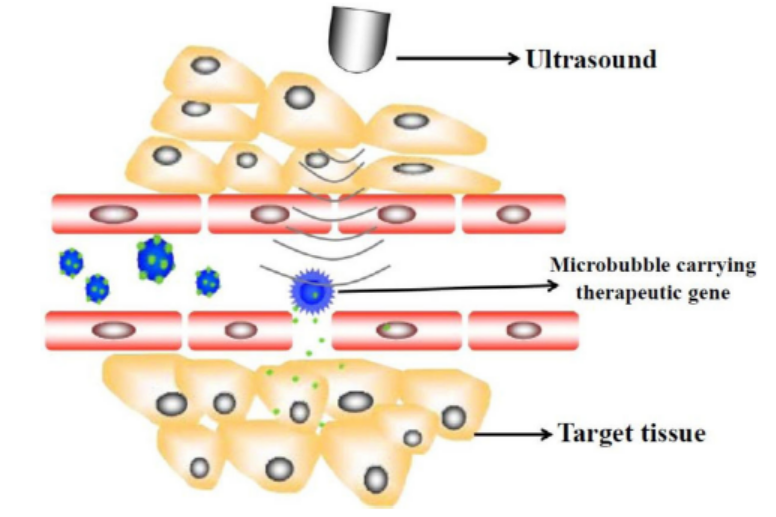
Figure 13.19: Ultrasound method
This method requires that a gene introduces DNA molecules into cells via acoustic
cavitation that temporarily changes the permeability of the cell membrane.
Sonoporation, or cellular sonication, is the use of sound (typically ultrasonic
frequencies) for modifying the permeability of the cell plasma membrane. This
technique is usually used in molecular biology and non-viral gene therapy in
order to allow uptake of large molecules such as DNA into the cell, in a cell
disruption process called transfection or transformation. Sonoporation employs
the acoustic cavitation of microbubbles to enhance delivery of these large molecules.
Sonoporation is under active study for the introduction of foreign genes in tissue
culture cells, especially mammalian cells. Sonoporation is also being studied for use
in targeted gene therapy in vivo, in a medical treatment scenario whereby a patient
is given modified DNA, and an ultrasonic transducer might target this modified DNAinto specific regions of the patient’s body.
h. Shock wave-mediated genetic transformation method
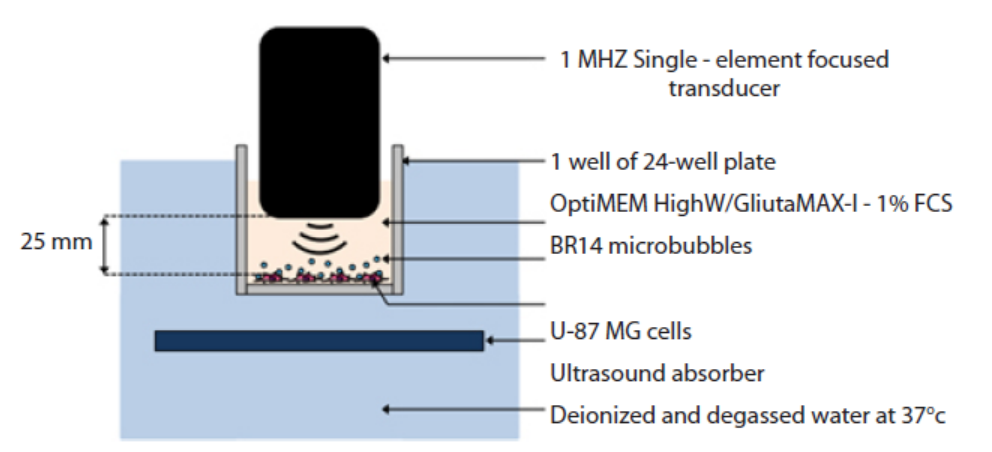
Figure 13.20: Shock wave-mediated genetic transformation method
This method involves sonoporation, based on the use of high frequency ultrasound
(1–10 MHz) in combination with gas microbubbles was introduced as a non-viral
physical method that is currently under evaluation for gene and drug delivery.
Sonoporation involves the treatment of a desired volume of cells in vitro or tissue in
vivo with ultrasound in the presence of microbubbles. These microbubbles, which
are formulated as lipid, albumin or polymer shelled micrometer sized gas bodies
in aqueous suspension, are commonly mixed with cells for in vitro applications or
administered by intravascular or intratissue injection for in vivo applications. The
exposure of microbubbles to ultrasound causes their periodic oscillations and/or
their collapse, under appropriate insonation conditions. It is now known that these
oscillations can induce micro-streaming, shock waves and/or micro-jets that can
affect the integrity of biological barriers (e.g. cell membrane, endothelial barrier). The
use of sonoporation to deliver therapeutic molecules to tissues has been extensively
explored over the past decade.
For example, the loco-regional delivery of anti-tumoral drugs has been reported
and is now under clinical investigation. Sonoporation has been successfully used to
transfer nucleic acids such as DNA into the heart, skeletal muscle, tumors, vessels,
liver and kidney. This method enables exogenous delivery of molecules with minimal
cell or tissue damage, inflammation and/or immunological response. In addition,
ultrasound can be non-invasively targeted to a specific volume of superficial tissues
or deeply embedded organs. Taken together, these properties make sonoporationan innovative and compelling method for gene and drug delivery.
Table 13.2: Summary of some physical methods for genetic transformation of cells

Application 13.4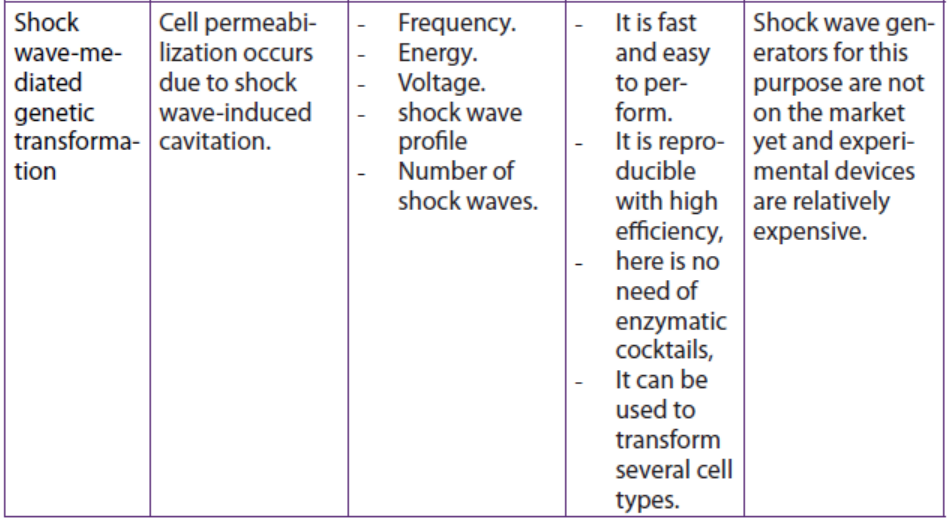
1. Observe the figure below and respond to the following questions.
a. Identify A and B.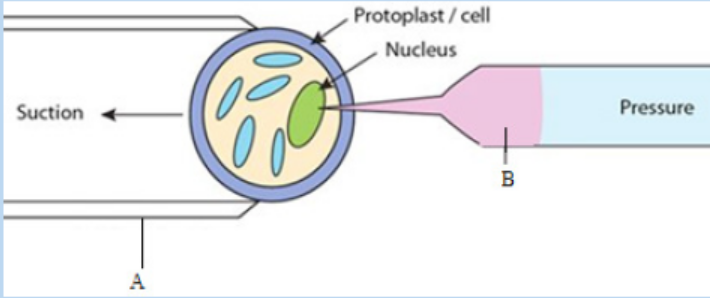
b. Describe briefly the method shown by this figure.
2. Distinguish between ultrasound technique and shock waves technique interms of involved parameters
13.5 Principles of Polymerase Chain Reaction (PCR) in cloningand amplifying DNA
Activity 13.5
Using different Biology textbooks, charts and computer simulations, discuss
the mechanism of artificial DNA synthesis focusing on Polymerase Chain
Reaction (PCR). During your discussion answer to the following questions:
1. Identify the types of artificial DNA synthesis.
2. Analyse the main steps of PCR3. Explain the role of Thermus aquaticus in PCR
Polymerase chain reaction is a technique that uses the enzyme called DNA
polymerase to produce millions of copies of a particular piece of DNA. For simplicity,the two original single strands are not shown after step 3. The main steps of the PCR:
– The DNA duplex is heated to 90 oC to separate the two strands (step 1:
denaturation).
– The mixture is cooled to 60 oC to allow the primers to anneal to their
complementary sequences (step 2: annealing).
– At 72 oC the primers direct the thermostable DNA polymerase to copy each ofthe template strands (step 3: extension or elongation of primers).
The three steps of the PCR are repeated many times to yield many thousands of
copies of the original target sequence. Genes can be cloned by cloning the bacterial
cells that contain them, but this requires quite a lot of DNA in the first place. PCR can
clone (or amplify) DNA samples as small as a single molecule. It is a newer technique,
having been developed in 1983 by Kary Mullis, for his discovery he won the Nobel
Prize in 1993.
The polymerase chain reaction is simply DNA replication in a test tube. If a length
of DNA is mixed with the four nucleotides (A, T, C and G) and the enzyme DNA
polymerase in a test tube, then the DNA will be replicated many times.
Normally, in vivo where DNA replication occurs, the DNA double helix would be
separated by the enzymes DNA gyrase and DNA helicase, but in PCR (in vitro) the
strands are separated by heating to 95°C for two minutes. This breaks the hydrogen
bonds. DNA polymerisation always requires short lengths of DNA (about 20 bases
pair long) called primers, to get it started. In vivo the primers are made during
replication by DNA polymerase, but in vitro they must be synthesised separately
and added at this stage. This means that a short length of the sequence of the DNA
must already be known, but it does have the advantage that only the part between
the primer sequences is replicated. The DNA must be cooled to 40°C to allow theprimers to anneal to their complementary sequences on the separated DNA strands.
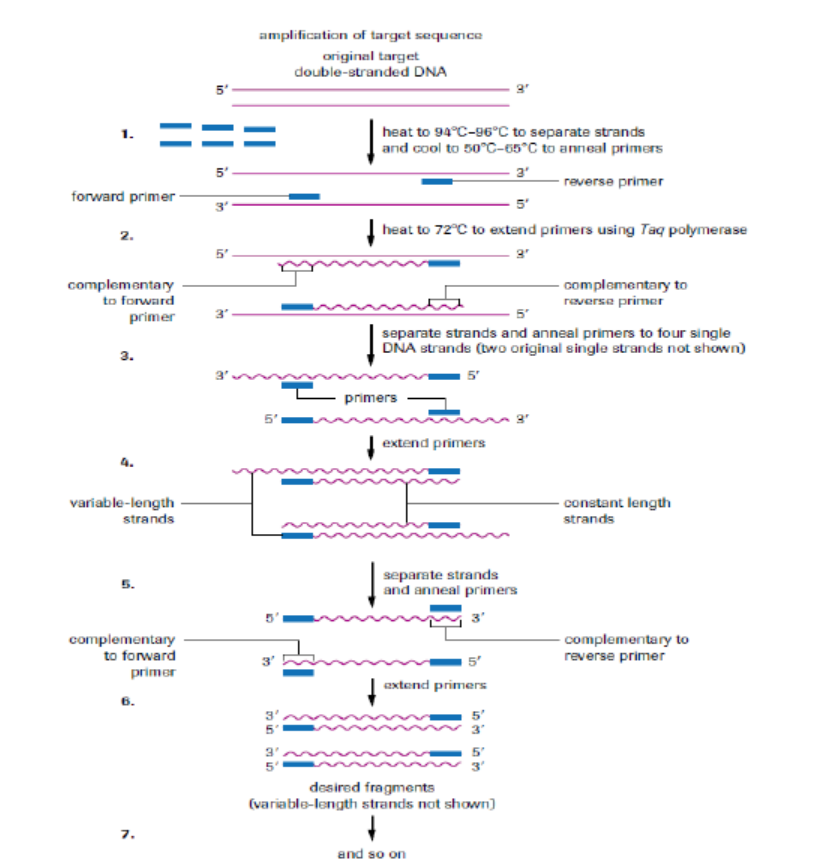
Figure 13.21: Polymerase chain reaction.
The enzyme (Taq polymerase) used in PCR is derived from the thermophilic bacterium
Thermus aquaticus, which grows naturally in hot springs at a temperature of 90°C, so
it is not denatured by the high temperatures in step 2. Its optimum temperature is
about 72°C, so the mixture is heated to this temperature for a few minutes to allow
replication to take place as quickly as possible. Once the primers have annealed, Taqpolymerase, a DNA polymerase, can build complementary strands using free
nucleotides that have been added to the solution. Each original DNA molecule has
now been replicated to form two molecules. The cycle is repeated from step 2 and
each time the number of DNA molecules doubles. This is why it is called a chain
reaction, since the number of molecules increases exponentially, like an explosivechain reaction. Typically PCR is run from 20 to30 cycles.
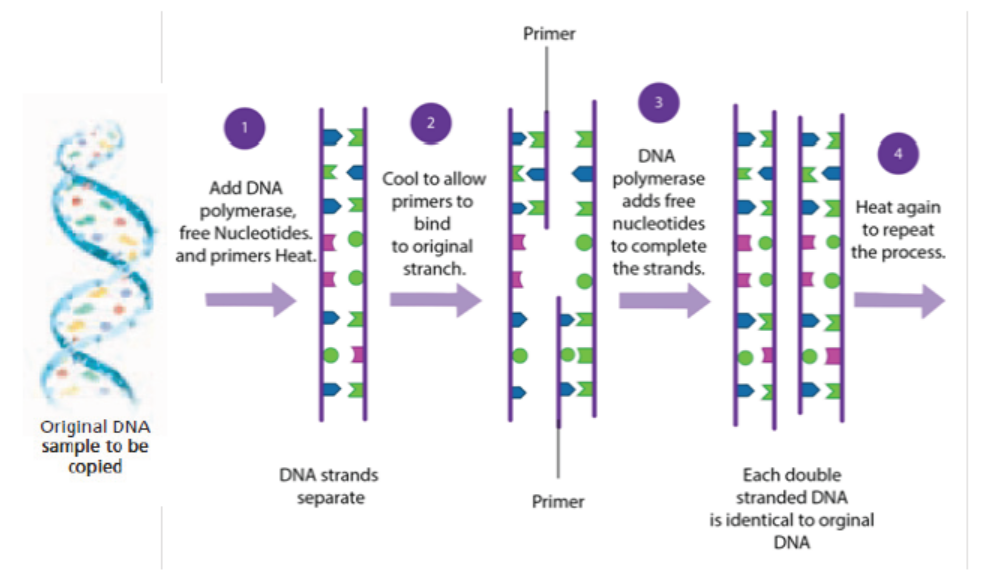
Figure 13.22: Summary of PCR technique
Note that:
Artificial DNA synthesis, sometimes known as DNA printing is a method
in synthetic biology that is used to create artificial genes in the laboratory. The types
of artificial DNA synthesis include recombinant DNA technology, gene purification
and PCR (Polymerase Chain Reaction). All of these types have been described aboveunder headings 13.1, 13.3 and 13.5 respectively.
Application 13.5
1. Explain briefly the artificial DNA printing2. Differentiate between PCR and DNA replication
13.6 Gel electrophoresis
Activity 13.6
Using computer animations and biology textbooks, observe the gel
electrophoresis used to analyse proteins and nucleic acids to distinguish
between alleles of a gene; then answer the following questions:
1. What is meant by gel electrophoresis?
2. Describe briefly the steps taken during separation of DNA, RNA fromthe mixture by use of gel electrophoresis.
Gel electrophoresis is a laboratory technique used to separate mixtures of DNA,
RNA or proteins according to molecular size. In gel electrophoresis, the molecules
to be separated are pushed by an electrical field through a gel that contains smallpores.
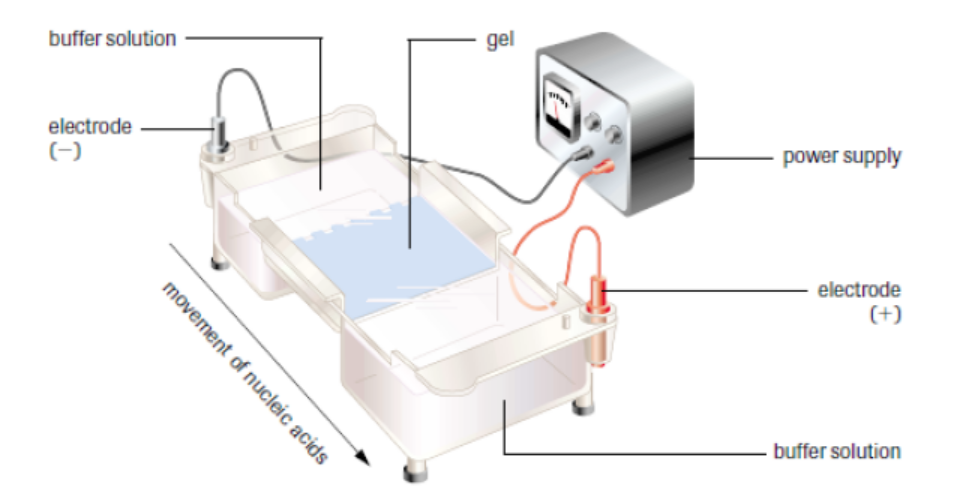
Figure 13.23: Setup of gel electrophoresis.
In a common gel electrophoresis setup, a nucleic acid such as DNA is loaded into
wells at one end of the gel and then migrates toward the positive electrode at the
opposite end. The rate of migration of fragments varies with size. The steps of gel
electrophoresis are shown below.
– The DNA samples are cut with a restriction enzyme into smaller segments of
various sizes. The DNA is then placed in wells made on a thick gel.
– An electric current runs through the gel for a given period of time. Negativelycharged DNA fragments migrate toward the positively charged end of the
porous gel. Smaller DNA fragments migrate faster and farther than longer
fragments, and this separates the fragments by size. The gel floats in a buffer
solution within a chamber between two electrodes.
– The DNA is transferred to a nylon membrane and radioactive probes are added.
The probes bind to complementary DNA.
– The X-ray film is exposed to the radiolabelled membrane. The resulting patternof bands is called a DNA fingerprint.
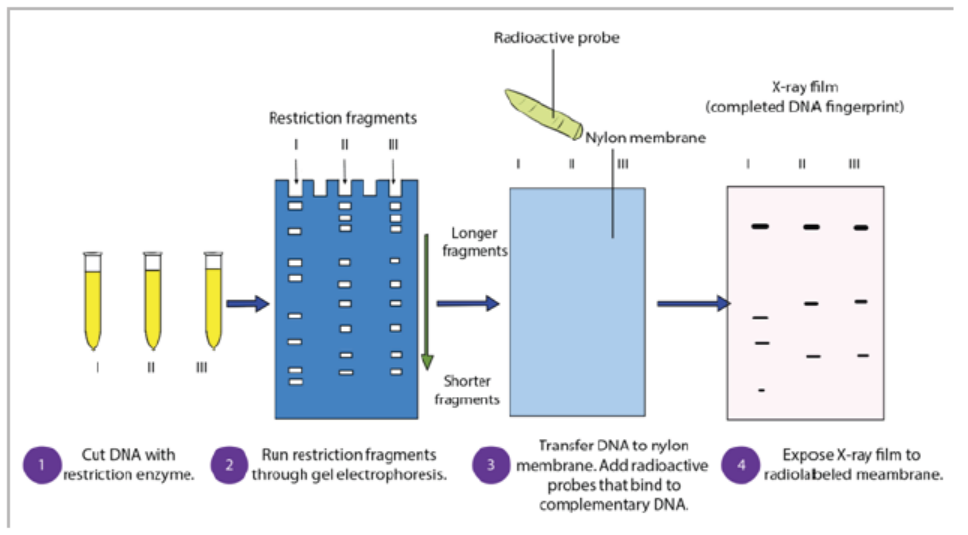
Figure 13.24: Steps of gel electrophoresis
During electrophoresis, DNA fragments migrate through the gel at a rate that is
inversely proportional to the logarithm of their size. The shorter the fragment is,
the faster it will travel because of its ability to navigate through the pores in the gel
more easily than a large fragment can. Larger fragments are hampered by their size.
Hence, the longer a nucleotide chain, the longer it takes for the migration.
Gel electrophoresis takes advantage of DNA’s negative charge. Using direct current,
a negative charge is placed at one end of the gel where the wells are, and a positive
charge is placed at the opposite end of the gel. The electrolyte solution conveys
the current through the gel. The negatively charged DNA will migrate toward the
positively charged electrode, with the shorter fragments migrating faster than the
longer fragments, achieving separation. Small molecules found within the loading
dye migrate ahead of all the DNA fragments. Since the small molecules can be
visualized, the electrical current can be turned off before they reach the end of thegel.
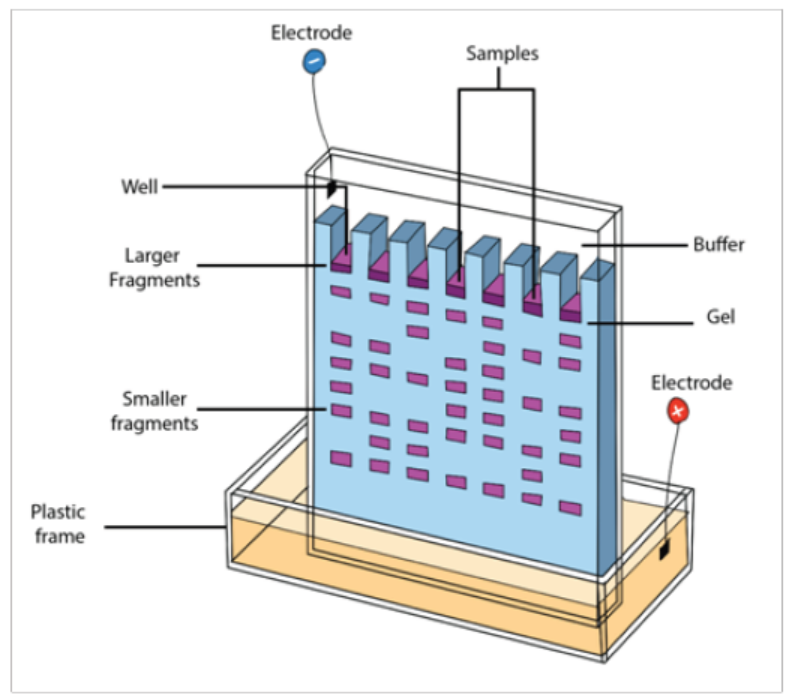
Figure 13.25: Fragments arrangement in gel electrophoresis
Once gel electrophoresis is complete, the DNA fragments are made visible by
staining the gel. The set of fragments generated with a particular restriction enzyme
produces a banding pattern characteristic for that DNA. The most commonly used
stain is ethidium bromide. Ethidium bromide is a flat molecule that fluoresces under
ultraviolet (UV) light and is able to insert itself among the rungs of the ladder of
DNA. When the gel is subjected to UV light, the bands of DNA are visualized because
the ethidium bromide is inserted among the nucleotides. The size of the fragments
is then determined using a molecular marker as a standard. The molecular marker,
which contains fragments of known size, is run under the same conditions (in thesame gel) as the digested DNA.
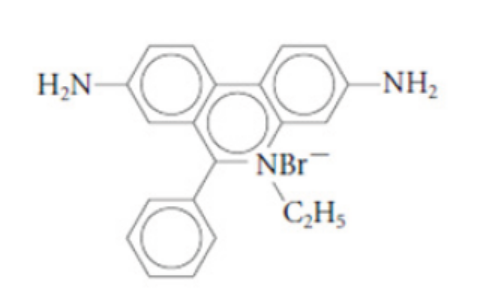
Figure 13.26: Ethidium bromide
Gel electrophoresis is not limited to the separation of nucleic acids but is also
commonly applied to proteins. Proteins are usually run on polyacrylamide gels,
which have smaller pores, because proteins are generally smaller in size than nucleic
acids. Proteins, however, are not negatively charged; thus, when researchers want to
separate proteins using gel electrophoresis, they must first mix the proteins with a
detergent called sodium dodecyl sulfate. This treatment makes the proteins unfold
into a linear shape and coats them with a negative charge, which allows them to
migrate toward the positive end of the gel and be separated. Finally, after the DNA,
RNA, or protein molecules have been separated using gel electrophoresis, bands
representing molecules of different sizes can be detected. The gel electrophoresis is
used for different purposes such as DNA analysis, protein and antibody interactions,testing antibiotics and testing vaccines.
Application 13.6
1. Identify the processes in which gel electrophoresis is used.
2. Describe the importance of restriction enzyme in gel electrophoresis.3. Explain the role of ethidium bromide in gel electrophoresis
13.7 Use of microarrays in the analysis of genomes and in detectingmRNA
Activity 13.7
Using different Biology textbooks, charts and internet; research information
about the use of microarrays in the analysis of genomes, answer to the
following questions.
1. What is microarray in genome analysis?2. Summarize the steps of microarray process in genome analysis.
DNA microarray, also commonly known as RNA chip or gene chip or biochip,
is technique consisting of a two-dimensional arrangement of DNA molecules
representing thousands of cloned genes on a solid surface such as a microscopic slide.
DNA microarray shows active genes that are being expression. Since a microarray
technology has the potential to examine the expression of several genes at a time,
it promises to revolutionize the way scientists study gene expression. For these
reasons, DNA microarrays are considered important tools for discovery in clinicalmedicine.
A basic protocol for a DNA microarray is as follows:
1. Isolate and purify mRNA from samples of interest:
As we are interested in comparing gene expression, one sample usually serves as
control, and another sample would be the experiment (for example, a healthy or
normal cell versus cancer cell).
2. Reverse transcribe and label the mRNA:
In order to detect the transcripts by hybridization, they need to be labeled, and
because starting material may be limited, an amplification step is also used. Labeling
usually involves performing a reverse transcription (RT) reaction to produce a
complementary DNA strand (cDNA) and incorporating a florescent dye that has
been linked to a DNA nucleotide, producing a fluorescent cDNA strand. Disease and
healthy samples can be labeled with different dyes and cohybridized onto the same
microarray in the following step. Some protocols do not label the cDNA but use a
second step of amplification, where the cDNA from RT step serves as a template toproduce a labeled cRNA strand.
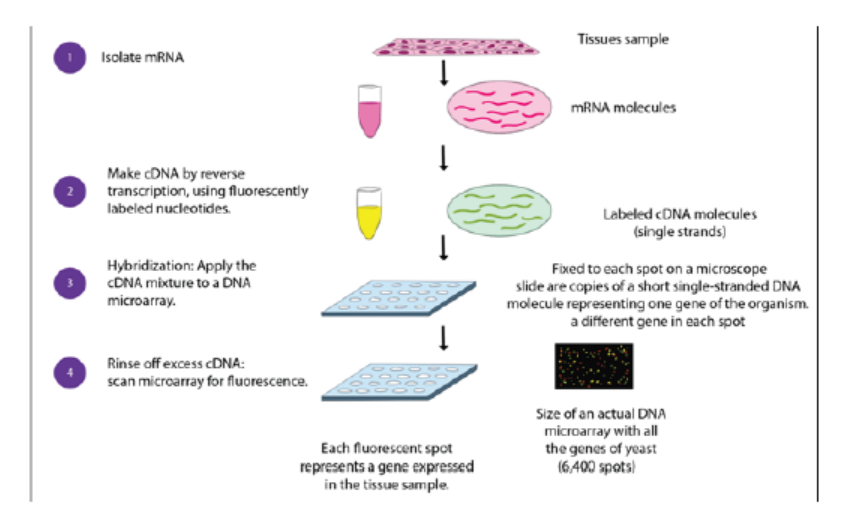
Figure 13.27: Procedure of microarray
3. Hybridize the labelled target to the microarray:
This step involves placing labelled cDNAs onto a DNA microarray where it will
hybridize to their synthetic complementary DNA probes attached on the microarray.
A series of washes are used to remove non-bound sequences. In molecular biology,
a hybridization probe is a fragment of DNA or RNA of variable length (usually 100–
1000 bases long) which can be radioactively labelled. It can then be used in DNA or
RNA samples to detect the presence of nucleotide sequences (the DNA target) thatare complementary to the sequence in the probe.
4. Scan the microarray and quantitate the signal:
The fluorescent tags on bound cDNA are excited by a laser and the fluorescently
labelled target sequences that bind to a probe generate a signal. The total strength
of the signal depends upon the amount of target sample binding to the probes
present on that spot. Thus, the amount of target sequence bound to each probe
correlates to the expression level of various genes expressed in the sample. Thesignals are detected, quantified, and used to create a digital image of the array.
Figure 13.28: A DNA microarray as viewed with a laser scanner. The colours are analysed to show which
genes or alleles are present
Application 13.7
1. Explain the role of reverse transcriptase in microarray technique
2. Describe the DNA probe
3. The figure below shows the microarray experiment in which the nucleicacids from cancer cells and normal cells are involved.
a. Redraw this figure identifying the steps X and Y and the molecule Z.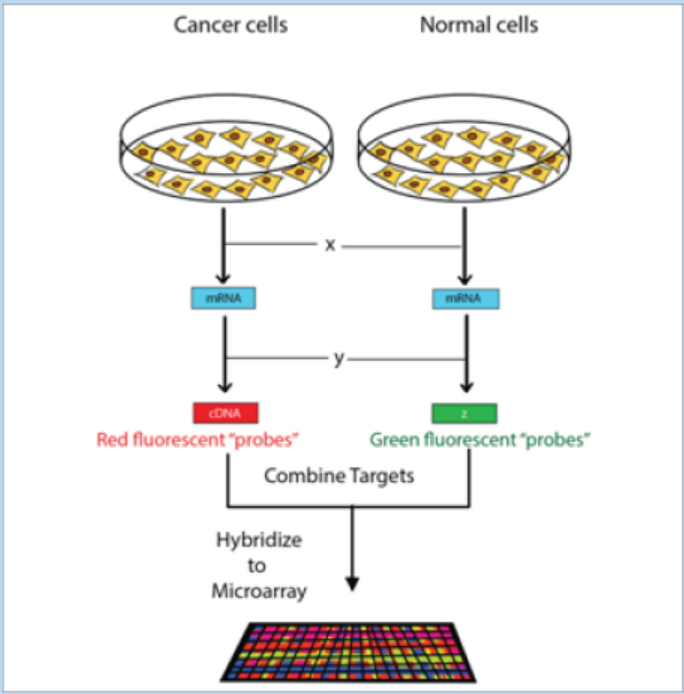
b. Describe briefly the aim of this experiment.
End of unit assessment 13
1. Choose the letter corresponding to the best answer.
(i) Different enzymes are used in the various steps involved in the production
of bacteria capable of synthesizing a human protein. Which step is
catalysed by a restriction enzyme?
a. Cloning DNA
b. Cutting open a plasmid vector
c. Producing cDNA from mRNA
d. Reforming the DNA double helix
(ii) What describes a promoter?
a. A length of DNA that controls the expression of a gene.
b. A piece of RNA that binds to DNA to switch off a gene.
c. A polypeptide that binds to DNA to switch on a gene
d. A triplet code of three DNA nucleotides that codes for ‘stop’
(iii) Which statement correctly describes the electrophoresis of DNA
fragments?
a. Larger fragments of DNA move more rapidly to the anode than smaller
fragments.
b. Positively charged fragments of DNA move to the anode.
c. Small negatively charged fragments of DNA move rapidly to the cathode.
d. Smaller fragments of DNA move more rapidly than larger fragments.
2. Explain the advantages of using plasmids as vectors.
3. The latest estimate of the number of genes in the human genome is 21 000.
Before the invention of microarrays, it was very time consuming to find out
which genes were expressed in any particular cell.
a. Explain how it is possible to find out which genes are active in a cell at a
particular time in its development.
b. Why is it not possible to use the same technique to find out which genes
are active in red blood cells?
4. Refer to what you have studied on DNA and PCR,
a. How many molecules of DNA are produced from one double-stranded
starting molecule, after eight cycles of PCR?
b. Explain why it is not possible to use PCR to increase the number of RNA
molecules in the same way as it is used to increase the number of DNAmolecules.
5. Complete the following table of the techniques used in gene technology.