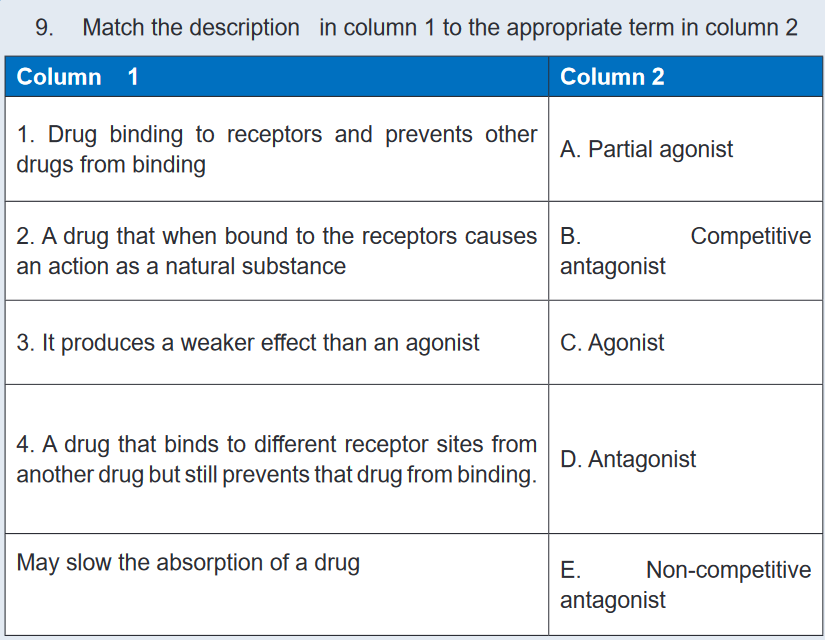
UNIT 2: PHARMACOKINETICS AND PHARMACODYNAMICS
Key Unit Competence
Explain the application of pharmacokinetcs and pharmacodynamics during clinical
practice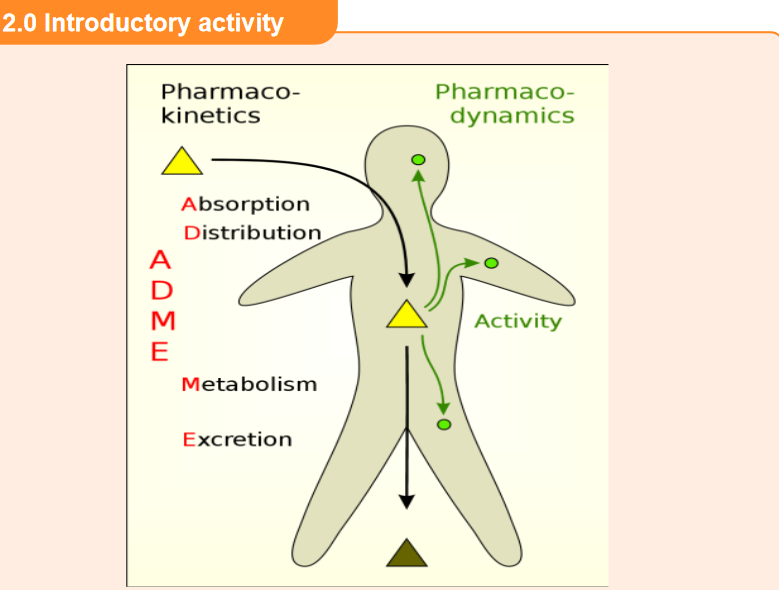
1. That image above represents a patient who has ingested a drug. What
do you think the arrows in the image indicate?
2. In your daily life, what do you think happens in the body after ingestion of
medications (tablets)?2.1 Introduction to Pharmacokinetics
Learning activity 2.1
You are placed at a health post in the clinical placement, and the patient consults
for his medical condition follow up. As he has a chronic disease, you inquire about
his health status, focusing on kidney function, bearing in mind that the drug is
eliminated via the urinary system. Then your colleague says that he heard that
pharmacokinetics of each needs to be taken into consideration while prescribing
a drug. He is curious, and would like to get more explanations from you.1. How can you briefly explain the word “pharmacokinetics” to your
colleague?2. Mention 4 phases/processes of pharmacokinetics.
CONTENT SUMMARY
Pharmacokinetics, sometimes described as what the body does to a drug, refers to
the movement of drug into, through, and out of the body.Pharmacokinetics involves the study of absorption, distribution, metabolism
(biotransformation), and excretion of drugs. In clinical practice, pharmacokinetic
considerations include the onset of drug action, drug half-life, timing of the peak
effect, duration of drug effects, metabolism or biotransformation of the drug, and
the site of excretion.Critical Concentration
After a drug is administered, its molecules first must be absorbed into the body;
then they make their way to the reactive tissues. If a drug is going to work properly
on these reactive tissues, and thereby have a therapeutic effect, it must attain a
sufficiently high concentration in the body. The amount of a drug that is needed to
cause a therapeutic effect is called the critical concentration.Drug evaluation studies determine the critical concentration required to cause
a desired therapeutic effect. The recommended dose of a drug is based on the
amount that must be given to eventually reach the critical concentration. Too much
of a drug will produce toxic (poisonous) effects, and too little will not produce the
desired therapeutic effects.Loading Dose
Some drugs may take a prolonged period to reach a critical concentration. If their
effects are needed quickly, a loading dose is recommended.Dynamic Equilibrium
The actual concentration that a drug reaches in the body results from a dynamic
equilibrium involving several processes:
1. Absorption from the site of entry
2. Distribution to the active site
3. Biotransformation (metabolism) in the liver
4. Excretion from the bodyThese processes are key elements in determining the amount of drug (dose)
and the frequency of dose repetition (scheduling) required to achieve the critical
concentration for the desired length of time. When administering a drug, the nurse
needs to consider the phases of pharmacokinetics so that the drug regimen can be
made as effective as possible.Self- assessment 2.1
1. There are some drugs that may take a prolonged period to reach a critical
concentration. If their effects are needed quickly, a maintenance dose is
recommended. (TRUE or FALSE)2. A right sequence of pharmacokinetic processes for a drug given by oral
route is:
A. Absorption, Distribution, Biotransformation and Excretion
B. Distribution, Absorption, Biotransformation and Excretion
C. Biotransformation, Absorption, Distribution, and Excretion
D. Excretion, Absorption, Distribution, and MetabolismSource: Library textbooks of pharmacology (Karch, A.M. (2013). Focus
on Nursing Pharmacology): On chapter of pharmacokinetics and
pharmacodynamics.2.2 Absorption of drugs
Learning activity 2.2
A patient X was received at the health post presenting severe respiratory disease.
An associate nurse student in clinical practice suggests administering a drug via
oral route but the nurse tells him to administer injectable drug rather than oral
drug, as the injectable form can work quickly.1. Referring to drug absorption, explain why the nurse preferred injectable
drug form.
2. List at least 3 factors that can affect absorption of drugs administered by
oral route.CONTENT SUMMARY
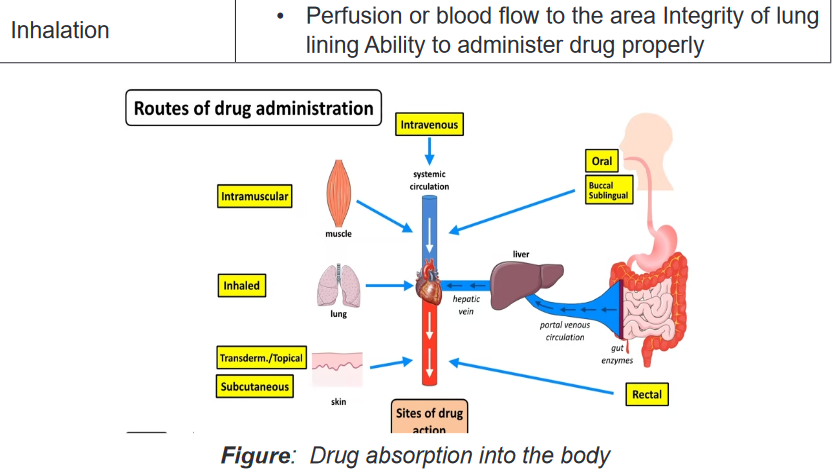
Absorption refers to what happens to a drug from the time it is introduced to the
body until it reaches the circulating fluids and tissues. Drugs can be absorbed
from many different areas in the body: through the GI tract either orally or rectally,
through mucous membranes, through the skin, through the lung, or through muscle
or subcutaneous tissues.Drug absorption is influenced by the route of administration. Generally, drugs given
by the oral route are absorbed more slowly than those given parentally. Of the
parenteral route, IV administered drugs are absorbed the fastest.Routes of Administration
The oral route is the most frequently used drug administration route in clinical
practice. Oral administration is not invasive, and, as a rule, oral administration is
less expensive than drug administration by other routes. It is also the safest way to
deliver drugs. Patients can easily continue their drug regimen at home when they
are taking oral medications. Oral administration subjects the drug to a number of
barriers aimed at destroying ingested foreign chemicals. The acidic environment of
the stomach is one of the first barriers to foreign chemicals.The acid breaks down many compounds and inactivates others. This fact is taken
into account by pharmaceutical companies when preparing drugs in capsule or
tablet form. The binders that are used often are designed to break down in ascertain
acidity and release the active drug to be absorbed.When food is present, stomach acidity is higher and the stomach empties more
slowly, thus exposing the drug to the acidic environment for a longer period. Certain
foods that increase stomach acidity, such as milk products, alcohol, and protein,
also speed the breakdown of many drugs.Other foods may chemically bind drugs or block their absorption. To decrease the
effects of this acid barrier and the direct effects of certain foods, oral drugs ideally
are to be given 1 hour before or 2 hours after a meal.Drugs that are injected IM (intramuscularly) are absorbed directly into the capillaries
in the muscle and sent into circulation. This takes time because the drug must
be picked up by the capillary and taken into the veins. Men have more vascular
muscles than women do. As a result, drugs administered to men via the IM route
reach a peak level faster than they do in women. Subcutaneous injections deposit
the drug just under the skin, where it is slowly absorbed into circulation. Timing of
absorption varies with subcutaneous injection, depending on the fat content of the
injection site and the state of local circulation.Absorption Processes: Drugs can be absorbed into cells through various
processes, which include passive diffusion and filtration.Passive diffusion is the major process through which drugs are absorbed into the
body. Passive diffusion occurs across a concentration gradient.When there is a greater concentration of drug on one side of a cell membrane,
the drug will move through the membrane to the area of lower concentration.
This process does not require any cellular energy. Unlike passive diffusion, active
transport is a process that uses energy to actively move a molecule across a cell
membrane. The molecule may be large, or it may be moving against a concentration
gradient. This process is not very important in the absorption of most drugs, but it is
often a very important process in drug excretion in the kidney.Filtration involves movement through pores in the cell membrane, either down a
concentration gradient or as a result of the pull of plasma proteins (when pushed
by hydrostatic, blood, or osmotic pressure). Filtration is another process the body
commonly uses in drug excretion.TABLE 2.2 Factors that affect absorption of drugs

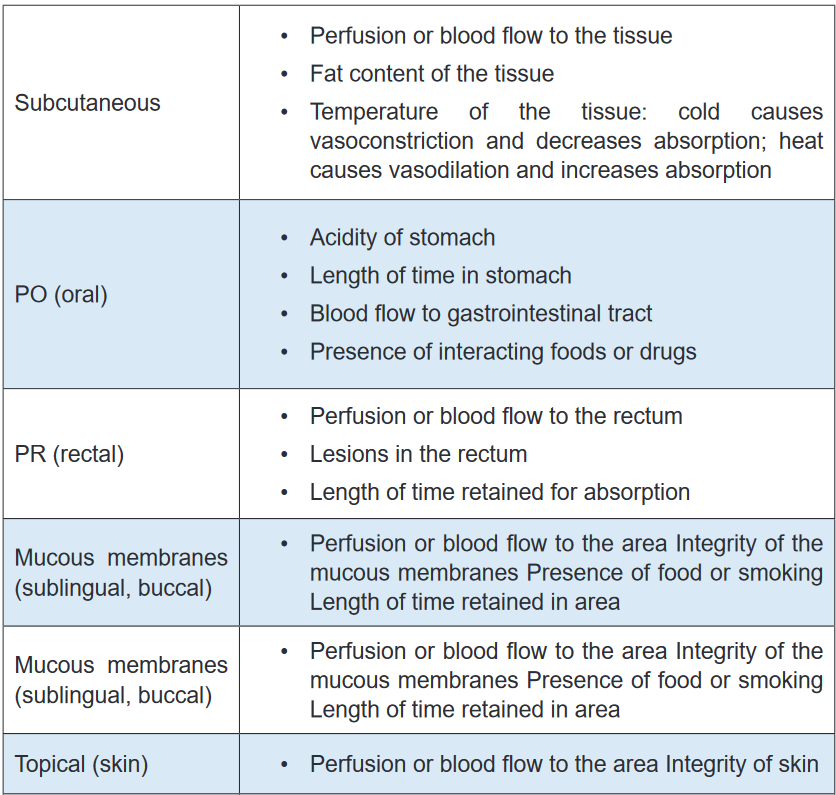
Self- assessment 2.2
Multiple choice questions
1. Which of the following drug transport ways requires energy in drug
movement in aqueous diffusion model?
A. Active transport
B. Facilitated transport
C. Passive transport
D. Filtration2. Identify the factors that can affect the absorption of the drugs administered
by the following routes:
A. IM (Intramuscularly)
B. SC (Subcutaneously)
C. IV (Intravenously)2.3 Distribution of drugs
Learning activity 2.3
Read the scenario below:
A 37-year-old female patient consults the health faciluty for her localized leg
infection. She is known as a diabetic for the last 10 years, and has developed
circulatory complications that affected different body parts including the lower
limbs. This infection can be treated by drugs that can act by either topical way or
systemic way. The nurse taking care of this patient is doubting the right mode to
use, and she wants to get advice from you as a student associate nurse carrying
out the clinical practice in her health post. You then advise him to choose the
drug that will work topically rather than the one that acts systemically.
1. Referring to the process of drug distribiution, why did you advise the
nurse to prescribe the drug that acts topically?2. Name 2 organs with high blood flow that are first to accumulate drugs
which are administered systemically?CONTENT SUMMARY
Once a drug has been absorbed from the stomach and/or intestines (GI Tract) into
the blood, it is circulated to some degree to all areas of the body to which there is
blood flow. This is the process of distribution. Organs with high blood flow, i.e.,
brain, heart, liver, etc. are the first to accumulate drugs, while connective tissue and
lesser-perfused organs are the last. The pattern of distribution of drug molecules by
different tissues after the chemical enters the circulatory system varies.Because of differences in pH, lipid content, cell membrane functions, and other
individual tissue factors, most drugs are not distributed equally in all parts of the
body. For example, the acidity of aspirin influences a distribution pattern that is
different from that of an alkaline product such as amphetamine. In same context,
tissue perfusion is a factor in treating a patient with diabetes who has a lower-leg
infection and needs antibiotics to destroy the bacteria in the area. In this case,
systemic drugs may not be effective because part of the disease process involves
changes in the vasculature and decreased blood flow to some areas, particularly
the lower limbs. If there is not adequate blood flow to the area, little antibiotic can be
delivered to the tissues, and little antibiotic effect will be seen. In addition, patients
in a cold environment may have constricted blood vessels (vasoconstriction) in the
extremities, which would prevent blood flow to those areas. The circulating blood
would be unable to deliver drugs to those areas, and the patient would receive little
therapeutic effect from drugs intended to react with those tissues.Many drugs are bound to plasma proteins such as albumin, and are not lipid soluble.
These drugs cannot be distributed to the central nervous system (CNS) because
of the effective blood–brain barrier (see later discussion), which is highly selective
in allowing lipid-soluble substances to pass into the CNS. Since only drugs that are
not bound are free to exert a pharmacologic effect, the ratio of “free” to “bound” drug
is important in determining the onset and duration of action of drugs. Highly bound
drugs are distributed less extensively throughout the body and are slower to act. By
virtue of their high binding to plasma proteins, they also stay in the body for longer
periods of time because the binding sites act as a sort of “reservoir” for the drug,
releasing drug molecules slowly.Protein Binding
Most drugs are bound to some extent to proteins in the blood to be carried into
circulation. The protein–drug complex is relatively large and cannot enter into
capillaries and then into tissues to react. The drug must be freed from the protein’s
binding site at the tissues.Some drugs are tightly bound and are released very slowly. These drugs have
a very long duration of action because they are not free to be broken down or
excreted. Therefore, they are released very slowly into the reactive tissue. Somedrugs are loosely bound; they tend to act quickly and to be excreted quickly. Some
drugs compete with each other for protein binding sites, altering effectiveness or
causing toxicity when the two drugs are given together.Blood–Brain Barrier
The blood–brain barrier is a protective system of cellular activity that keeps many
things (e.g., foreign invaders, poisons) away from the CNS. Drugs that are highly
lipid soluble are more likely to pass through the blood–brain barrier and reach the
CNS. Drugs that are not lipid soluble are not able to pass the blood–brain barrier.
This is clinically significant in treating a brain infection with antibiotics. Almost all
antibiotics are not lipid soluble and cannot cross the blood–brain barrier. Effective
antibiotic treatment can occur only when the infection is severe enough to alter the
blood–brain barrier and allow antibiotics to cross.Although many drugs can cause adverse CNS effects, these are often the result
of indirect drug effects and not the actual reaction of the drug with CNS tissue.
For example, alterations in glucose levels and electrolyte changes can interfere
with nerve functioning and produce CNS effects such as dizziness, confusion, or
changes in thinking ability.Placenta and Breast Milk
Many drugs readily pass through the placenta and affect the developing fetus in
pregnant women. As it has been approved, it is best not to administer any drugs to
pregnant women because of the possible risk to the fetus. Drugs should be given
only when the benefit clearly outweighs any risk. Many other drugs are secreted
into breast milk and therefore have the potential to affect the neonate. Because of
this possibility, the nurse must always check the ability of a drug to pass into breast
milk when giving a drug to a breast-feeding mother.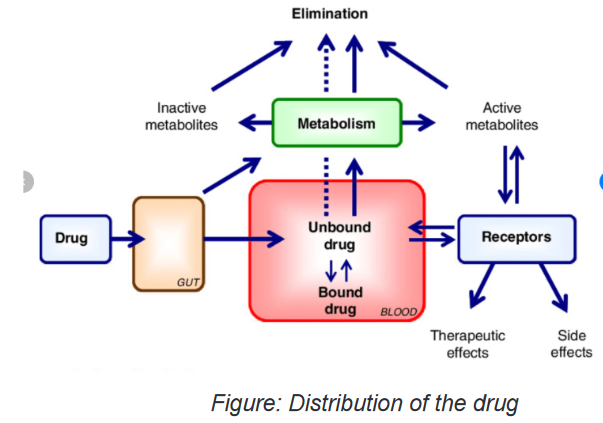
Self- assessment 2.3
1. After absorption of the drug from the stomach and/or intestines (GI Tract)
into the blood, the next pharmacokinetic step is:
A. Absorption
B. Excretion
C. Distribution
D. Metabolism2. During drug distribution, the drugs are bound to which of the following?
A. Proteins in the blood
B. Lipids in blood
C. Vitamins in blood
D. Minerals in blood2.4 Metabolism (Biotransformation) of drugs
Learning activity 2.4
Read the case study below and answer the questions related to it:
An 85-year-old male patient consults the health facility where you are placed
as an associate nurse during the clinical placement. He also suffers from a a
chronic liver disease, and he was prescribed the drugs that are metabolised in
the liver. You then advise the prescribing team to inform the patient that as he is
taking the drug, they need to advise adjust the dose and ensure that the patient
comes for follow up at the health facility.1. Referring to the metabolism of drugs, why did you advise the nurse to
adjust the dose and follow the client up?2. Name the main organ that is involved in metabolism of drugs.
CONTENT SUMMARY
Drugs in the blood and tissues must be inactivated and excreted from the body.
This process is initiated by altering the chemical structure of the drug in such a
way as to promote its excretion. The body is well prepared to deal with a myriad
of foreign chemicals. Enzymes in the liver, in many cells, in the lining of the GI
tract, and even circulating in the body detoxify foreign chemicals to protect the
fragile homeostasis that keeps the body functioning. The transformation of the
drug molecule into a chemically related substance that is more easily excretedfrom the body is called metabolism, biotransformation or detoxification. Drug
metabolism is the process by which the body breaks down and converts medication
into active chemical substances. Drugs can interact with other drugs, foods, and
beverages. Interactions can lessen or magnify the desired therapeutic effect of a
drug, or may cause unwanted or unexpected side effects.Exogenous compounds (xenobiotics) must be metabolized before they can be
excreted. The biochemical transformation of xenobiotics, such as alcohol, nicotine
and drugs is a prime activity of the liver. In addition to the liver, biotransformation
processes occur in plasma, in the lungs, in the gastrointestinal tract and in the
skin. The liver is the organ that plays a major role in metabolism, digestion,
detoxification, and elimination of substances from the body. Think of the liver as a
sewage treatment plant. Everything that is absorbed from the GI tract first enters
the liver to be “treated.” The liver detoxifies many chemicals and uses others to
produce needed enzymes and structures. Enzymes in the liver are responsible
for chemically changing drug components into substances known as metabolites.
Metabolites are then bound to other substances for excretion through the lungs, or
bodily fluids such as saliva, sweat, breast milk, and urine, or through reabsorption
by the intestines. The metabolic rate can vary significantly from person to person,
and drug dosages that work quickly and effectively in one individual may not work
well for another.First-Pass Effect
Drugs that are taken orally are usually absorbed from the small intestine directly
into the portal venous system (the blood vessels that flow through the liver on
their way back to the heart). Aspirin and alcohol are two drugs that are known to
be absorbed from the lower end of the stomach. The portal veins deliver these
absorbed molecules into the liver, which immediately transforms most of the
chemicals delivered to it by a series of liver enzymes. These enzymes break the
drug into metabolites, some of which are active and cause effects in the body,
and some of which are deactivated and can be readily excreted from the body. As
a result, a large percentage of the oral dose is destroyed at this point and never
reaches the tissues. This phenomenon is known as the first-pass effect. The portion
of the drug that gets through the first pass effect is delivered to the circulatory
system for transport throughout the body.Injected drugs and drugs absorbed from sites other than the GI tract undergo a
similar biotransformation when they pass through the liver. Because some of the
active drug already has had a chance to reach the reactive tissues before reaching
the liver, the injected drug is often more effective at a lower dose than the oral
equivalent. Thus, the recommended dose for oral drugs can be considerably higher
than the recommended dose for parenteral drugs, taking the first-pass effect into
accountFactors that influence drug metabolism
These include:
• Genetics,
• Environment,
• Nutrition, and
• Age. Infants and elderly patients may have a reduced capacity to metabolize
certain drugs, and may require adjustments in dosage.Self- assessment 2.4
1. Which of the following factors may impact negatively the drug metabolism?
A. Proper nutrition
B. Advanced age (elderly)
C. Healthy liver
D. Healthy young person2. Which of the following routes of drug administration would be more likely
to make the drug subject to first-pass effect?
A. Oral
B. Intravenous
C. Intraarterial
D. Intranasal2.5 Excretion of drugs
Learning activity 2.5
A patient known for chronic heart failure consults hospital for the appointment.
The doctor decides to test kidney function and finds the client has developed
also kidney failure. The doctor prescribes a drug eliminated by the kidneys, but
reduces the dose. The client asks the doctor why to reduce the dose.
1. Referring to the excretion of the drug, what should the doctor tell the
client?CONTENT SUMMARY
Excretion is the removal of a drug from the body. The skin, saliva, lungs, bile, and
feces are some of the routes used to excrete drugs. The kidneys, however, play the
most important role in drug excretion. Drugs that have been made water soluble
in the liver are often readily excreted from the kidney by glomerular filtration (the
passage of water and water-soluble components from the plasma into the renal
tubule).Other drugs are secreted or reabsorbed through the renal tubule by active transport
systems. The active transport systems that move the drug into the tubule often do
so by exchanging it for acid or bicarbonate molecules. Therefore, the acidity of
urine can play an important role in drug excretion.This concept is important to remember when trying to clear a drug rapidly from
the system or trying to understand why a drug is being given at the usual dose but
is reaching toxic levels in the system. One should always consider the patient’s
kidney function and urine acidity before administering a drug. Kidney dysfunction
can lead to toxic levels of a drug in the body because the drug cannot be excreted.Half-Life
The half-life of a drug is the time it takes for the amount of drug in the body to
decrease to one half of the peak level it previously achieved. For instance, if a
patient takes 20 mg of a drug with a half-life of 2 hours, 10 mg of the drug will
remain 2 hours after administration. Two hours later, 5 mg will be left (one half of
the previous level); in 2 more hours, only 2.5 mg will remain. This information is
important in determining the appropriate timing for a drug dose or determining the
duration of a drug’s effect on the body.The absorption rate, the distribution to the tissues, the speed of biotransformation,
and how fast a drug is excreted are all taken into consideration when determining
the half-life of the drug. The half-life that is indicated in any drug monograph is the
half-life for a healthy person.Using this information, one can estimate the half-life of a drug for a patient with
kidney or liver dysfunction (which could prolong the biotransformation and the time
required for excretion of a drug), allowing the prescriber to make changes in the
dosing schedule.The timing of drug administration is important to achieve the most effective drug
therapy. Nurses can use their knowledge of drug half-life to explain the importance
of following a schedule of drug administration in the hospital or at home.Self- assessment 2.5
1. The patient took 50 mg of drug with half-life of 2 hours at 8h00 AM. How
many mgs will be remaining in the body at 12h00 PM?
A. 25 mg
B. 20mg
C. 12.5mg
D. 6.25mg2. Define what half-life is.
3. In the following organs, which one plays the most important role in
excretion of a drug?
A. The skin
B. Saliva
C. Lungs
D. Kidney2.6 Factors influencing drug effects
Learning activity 2.6
A patient is brought to the health post where you are placed as a student associate
nurse, and you need to prescribe drugs for him.
It is a stunted kid who is brought by his parents, and he is aged 12 months.
1. Which factors influencing drug effects should you bear in mind for this
specific patient?2. Is it necessary to bear in mind factors that influence drug effects during
its prescription?CONTENT SUMMARY
When administering a drug to a patient, the nurse must be aware that the human
factor has a tremendous influence on what actually happens to a drug when it
enters the body. No two people react in exactly the same way to any given drug.
Even though textbooks and drug guides explain the pharmacodynamics and
pharmacokinetics of a drug, it must be remembered that such information usually is
based on studies of healthy adult males. Things may be very different in the clinical
setting. Consequently, before administering any drug, the nurse must consider a
number of factors influencing drug effects as follows:Weight
The recommended dose of a drug is based on drug evaluation studies and is
targeted at a 150-pound (around 70 kilos) person. People who are much heavier
may require larger doses to get a therapeutic effect from a drug because they have
increased tissues to perfuse and increased receptor sites in some reactive tissue.
People who weigh less than the norm may require smaller doses of a drug. Toxic
effects may occur at the recommended dose if the person is very small.
Age
Age is a factor primarily in children and older adults. Children are not just little adults.
Children metabolize many drugs differently than adults do, and they have immature
systems for handling drugs. Many drugs come with recommended pediatric doses,
and others can be converted to pediatric doses using one of several conversion
formulas.
Older adults undergo many physical changes that are a part of the aging process.
Their bodies may respond very differently in all aspects of pharmacokinetics—less
effective absorption, less efficient distribution because of fewer plasma proteins and
less efficient perfusion, altered biotransformation or metabolism of drugs because
of age-related liver changes, and less effective excretion owing to less efficient
kidneys. Many drugs now come with recommended doses for patients who are
older. The doses of other drugs also may need to be decreased for the older adult.
When administering drugs to a patient at either end of the age spectrum, one should
monitor the patient closely for the desired effects. If the effects are not what would
normally be expected, one should consider the need for a dose adjustment.Gender
Physiological differences between men and women can influence a drug’s effect.
When giving IM injections, for example, it is important to remember that men have
more vascular muscles, so the effects of the drug will be seen sooner in men than
in women. Women have more fat cells than men do, so drugs that deposit in fat
may be slowly released and cause effects for a prolonged period. For example,
gas anesthetics have an affinity for depositing in fat and can cause drowsiness
and sedation sometimes weeks after surgery. Women who are given any drug
should always be questioned about the possibility of pregnancy because, as stated
previously, the use of drugs in pregnant women is not recommended unless the
benefit clearly outweighs the potential risk to the fetus.Physiological Factors
Physiological differences such as diurnal rhythm of the nervous and endocrine
systems, acid–base balance, hydration, and electrolyte balance can affect the waythat a drug works on the body and the way that the body handles the drug. If a drug
does not produce the desired effect, one should review the patient’s acid–base and
electrolyte profiles and the timing of the drug.Pathological Factors
Drugs are usually used to treat disease or pathology. However, the disease that the
drug is intended to treat can change the functioning of the chemical reactions within
the body and thus change the response to the drug. Other pathological conditions
can change the basic pharmacokinetics of a drug. For example, GI disorders can
affect the absorption of many oral drugs. Vascular diseases and low blood pressure
alter the distribution of drug, preventing it from being delivered to the reactive tissue,
thus rendering the drug nontherapeutic. Liver or kidney diseases affect the way that
a drug is biotransformed and excreted and can lead to toxic reactions when the
usual dose is given.Genetic Factors
Genetic differences can sometimes explain patients’ varied responses to a given
drug. Some people lack certain enzyme systems necessary for metabolizing a drug,
whereas others have overactive enzyme systems that cause drugs to be broken
down more quickly. Still others have differing metabolisms or slightly different
enzymatic makeups that alter their chemical reactions and the effects of a given
drug.Immunological Factors
People can develop an allergy to a drug. After exposure to its proteins, a person
can develop antibodies to a drug. With future exposure to the same drug, that
person may experience a full-blown allergic reaction. Sensitivity to a drug can
range from mild (e.g., dermatological reactions such as a rash) to more severe
(e.g., anaphylaxis, shock, and death).Psychological Factors
The patient’s attitude about a drug has been shown to have an effect on how that
drug works. A drug is more likely to be effective if the patient thinks it will work than
if the patient believes it will not work. This is called the placebo effect.The patient’s personality also influences compliance with the drug regimen. Some
people who believe that they can influence their health actively seek health care
and willingly follow a prescribed regimen. These people usually trust the medical
system and believe that their efforts will be positive. Other people do not trust
the medical system. They may believe that they have no control over their health
and may be unwilling to comply with any prescribed therapy. Knowing a patient’s
healthseeking history and feelings about health care is important in planning aneducational program that will work for that patient. It is also important to know this
information when arranging for necessary follow-up procedures and evaluations. As
the caregiver most often involved in drug administration, the nurse is in a position
to influence the patient’s attitude about drug effectiveness. Frequently, the nurse’s
positive attitude, combined with additional comfort measures, can improve the
patient’s response to a medication.Environmental Factors
The environment can affect the success of drug therapy. Some drug effects are
enhanced by a quiet, cool, non-stimulating environment. For example, sedating
drugs are given to help a patient relax or to decrease tension. Reducing external
stimuli to decrease tension and stimulation help the drug be more effective. Other
drug effects may be influenced by temperature. For example, antihypertensives
that work well during cold, winter months may become too effective in warmer
environments, when natural vasodilation may lead to a release of heat that tends to
lower the blood pressure. If a patient’s response to a medication is not as expected,
look for possible changes in environmental conditions.Tolerance
The body may develop a tolerance to some drugs over time. Tolerance may arise
because of increased biotransformation of the drug, increased resistance to its
effects, or other pharmacokinetic factors. When tolerance occurs, the drug no long
causes the same reaction. Therefore, increasingly larger doses are needed to
achieve a therapeutic effect. An example is morphine, an opiate used for pain relief.
The longer morphine is taken, the more tolerant the body becomes to the drug, so
that larger and larger doses are needed to relieve pain. Clinically, this situation can
be avoided by giving the drug in smaller doses or in combination with other drugs
that may also relieve pain. Cross-tolerance—or resistance to drugs within the same
class—may also occur in some situations.Interactions
When two or more drugs or substances are taken together, there is a possibility
that an interaction can occur, causing unanticipated effects in the body. Alternative
therapies, such as herbal products, act as drugs in the body and can cause these
same interactions. Certain foods can interact with drugs in much the same way.
Usually this is an increase or decrease in the desired therapeutic effect of one or all
of the drugs or an increase in adverse effects.Self- assessment 2.6
1. List at least 5 factors that influence drug effects.
2. What is the ideal adult weight is considered while prescribing drugs?2.7 Drug-drug interactions
Learning activity 2.7
Read the case study below and answer the question related to it:
A 40-year-old male patient was prescribed a penicillin G injection, and he is
receiving concurrently tetracyclines taken by oral route. The symptoms of the
disease for which penicillin G was given persisted after 5 days of the treatment,
and the patient came back to the health facility where the drug was prescribed.
The prescribing personnel decide that both penicillin G and tetracyclines are
needed for this patient, and decides to increase the dose of penicillin G. After
2 days, the symptoms start to resolve until they completely disappear and the
patient improves.1. What do you think happened for this patient so that he did not improve
with the first period, and improved after increasing the dose of penicillin G?CONTENT SUMMARY
A drug-drug reaction is when there’s an interaction between two or more prescription
drugs. This can cause the medication to be less or more potent than intended or
result in unexpected side effects.Clinically significant drug-drug interactions occur with drugs that have small margins
of safety. If there is very little difference between a therapeutic dose and a toxic dose
of the drug, interference with the drug’s pharmacokinetics or pharmacodynamics
can produce serious problems. For example, drug-drug interactions can occur in
the following situations:At the site of absorption: One drug prevents or accelerates absorption of the
other drug. For example, the antibiotic tetracycline is not absorbed from the GI tract
if calcium or calcium products (milk) are present in the stomach.During distribution: One drug competes for the protein-binding site of another
drug, so the second drug cannot be transported to the reactive tissue. For example,
aspirin competes with the drug methotrexate for protein-binding sites. Because
aspirin is more competitive for the sites, the methotrexate is bumped off, resulting
in increased release of methotrexate and increased toxicity to the tissues.During biotransformation: One drug stimulates or blocks the metabolism of
the other drug. For example, warfarin (Coumadin), an oral anticoagulant, is
biotransformed more quickly if it is taken at the same time as barbiturates, rifampin,
or many other drugs. Because the warfarin is biotransformed to an inactive state
more quickly, higher doses will be needed to achieve the desired effect.During excretion: One drug competes for excretion with the other drug, leading
to accumulation and toxic effects of one of the drugs. For example, digoxin and
quinidine are both excreted from the same sites in the kidney. If they are given
together, the quinidine is more competitive for these sites and is excreted, resulting
in increased serum levels of digoxin, which cannot be excreted.At the site of action: One drug may be an antagonist of the other drug or may cause
effects that oppose those of the other drug, leading to no therapeutic effect. This is
seen, for example, when an antihypertensive drug is taken with an antiallergy drug
that also increases blood pressure. The effects on blood pressure are negated, and
there is a loss of the antihypertensive effectiveness of the drug.
If a patient is taking antidiabetic medication and also takes the herb ginseng, which
lowers blood glucose levels, he or she may experience episodes of hypoglycemia
and loss of blood glucose control.
Whenever two or more drugs are being given together, first consult a drug guide
for a listing of clinically significant drug-drug interactions. Sometimes problems
can be avoided by staggering the administration of the drugs or adjusting their
doses. For example, when penicillin G and tetracyclines are taken concurrently, the
effectiveness of penicillin G decreases. If this combination is used, the dose of the
penicillin should be increased.Drug-nonprescription treatment interaction refers the reaction between a drug
and a nonprescription treatment. These include over-the-counter (OTC) medications,
herbs, vitamins, or other supplements. An example of this type of interaction can
occur between a diuretic, a drug that attempts to rid the body of excess water and
salt taken with ibuprofen, as an non steroid anti-inflammatory drug. The ibuprofen
may reduce the diuretic’s effectiveness because ibuprofen often causes the body
to retain salt and fluid.Self- assessment 2.7
1. Referring to the lesson on drug-drug interactions, list the stages/sites at
which drug-drug interactions may happen.2. What do you understand by “drug-nonprescription treatment interaction”?
2.8 Drug- food/beverage interactions
Learning activity 2.8
our relative consulted the health post complaining of the low abdominal pain
and she has been prescribed medications. She was then told that she could not
take grapefruit juice while she is taking a drug but she does not understand why.
When she arrives home she asks you to give more explanation about why she
was requested not to take the grapefruit juice.
1. With reference to the interactions between drugs and food or beverages,
what will you tell to your sister?2. Drug-food/beverage interactions always result in decreased serum levels
of the concerned drugs. TRUE or FALSECONTENT SUMMARY
For the most part, a drug-food interaction occurs when the drug and the food are in
direct contact in the stomach. Some foods increase acid production, speeding the
breakdown of the drug molecule and preventing absorption and distribution of the
drug. Some foods chemically react with certain drugs and prevent their absorption
into the body. The antibiotic tetracycline cannot be taken with iron products for this
reason. Tetracycline also binds with calcium to some extent and should not be
taken with foods or other drugs containing calcium.Grapefruit juice has been found to affect liver enzyme systems for up to 48 hours
after it has been ingested. This can result in increased or decreased serum levels of
certain drugs. Many drugs come with the warning that they should not be combined
with grapefruit juice. This drug–food interaction does not take place in the stomach,
so the grapefruit juice needs to be avoided the entire time the drug is being used,
not just while the drug is in the stomach.In most cases, oral drugs are best taken on an empty stomach. If the patient cannot
tolerate the drug on an empty stomach, the food selected for ingestion with the drug
should be something that is known not to interact with it. Drug monographs usually
list important drug-food interactions and give guidelines for avoiding problems and
optimizing the drug’s therapeutic effects.Self- assessment 2.8
A patient consults the health post for pain during urination. He was then prescribed
antibiotic drugs. The associate nurse student in pharmacy of the health post
dispenses the medications but indicates the patient that he needs to take drugs
on the empty stomach.
1. Explain why it is better to take oral drugs on an empty stomach.2. Drug-food/beverage interactions occur for drugs administered orally only.
TRUE or FALSE2.9 Time-Response Relationships: Drug Plasma Levels
Learning activity 2.9
An associate nurse prescribed the drug to the client to be taken in equal intervals
of 4 hours a day. The first dose meant to achieve the target concentration rapidly
has to be taken at the time he consults the health post at 3h00 PM. The following
doses must then follow the first dose later on., respecting the intervals This
means that the second dose should be taken at 9.00 PM, third dose at 11 h00
PM. The client tells the associate nurse that he is going to take 2nd dose and 3
doses at the same time because he goes to bed at 8 h00’.
1. What should the associate nurse should the patient to understand the
reason of respecting the doses interval?2. Differentiate the loading dose from the maintenance dose.
CONTENT SUMMARY
Drugs are used for the treatment of diseases but the modes of administration
of drugs are different. The mode of administration is designed on the basis of
absorption, distribution, metabolism and excretion (ADME) of drugs. Drugs usually
follow two processes for their pharmacokinetic behaviour in the body. These are
first order and zero order processes.First order kinetic: This is the most common process for many drugs. The rate at
which absorption, distribution, metabolism and excretion occur are proportional to
the concentration of drugs i.e. constant fraction of this drug in the body disappears
in each equal interval of time.Zero order kinetic: It is independent of the amount of drug present at the particular
sites of drug absorption or elimination. Few drugs follow this process e.g. ethanol,
phenytoin. Here constant amount of the drug is eliminated in each equal interval of
time. On repeated administration of drug after certain stage it goes on accumulating
in the body and leads to toxic reactions.Steady state plasma concentration: When a drug dose is given repeatedly over
a given period, a steady state is eventually reached, at which point the amount of
drug absorbed is in equilibrium with that eliminated from the body. Steady state is
achieved after 4 to 5 half –lives for most of the drugs which follow first order kinetics.
For example, a drug with half-life of 6 hours will be expected to be at steady state
after more than 24 hours of administration. The pattern of drug accumulation during
repeated administration of drug at intervals equal to its elimination half-life.For some drugs, the effects are difficult to measure, toxicity and lack of efficacy
are both potential dangers, and/or the therapeutic window is narrow. In these
circumstances, doses must be adjusted carefully to a desired steady-state
concentration by giving loading and maintenance dosesLoading dose: The loading dose is one or a series of doses that may be given at
the onset of therapy with the aim of achieving the target concentration rapidly.Maintenance dose: To maintain the chosen steady-state or target concentration,
the rate of drug administration is adjusted such that the rate of input equals to rate
of loss.Self- assessment 2.9
Define the following terms
1. Steady state plasma concentration
2. Zero order kinetic
3. First order kinetic2.10 Introduction to pharmacodynamics
Learning activity 2.10
You receive a patient who consults the health post where you are placed as an
associate-nurse student. The patient consults for the difficulty swallowing and
fever. On the examination, you realize he has tonsillitis and you wish to prescribe
the drug. Before proceeding, you bear in your mind that the selective toxicity of
a drug must be considered always when the drug is being used.
1. What do you understand by the term “selective toxicity” as it is applied to
pharmacodynamics?2. How do we call the specific areas on cell membranes where many drugs
are thought to act?CONTENT SUMMARY
Pharmacodynamics is the study of the interactions between the chemical
components of living systems and the foreign chemicals, including drugs that enter
those systems. All living organisms function by a series of complicated, continual
chemical reactions. When a new chemical enters the system, multiple changes in
and interferences with cell functioning may occur. To avoid such problems, drug
development works to provide the most effective and least toxic chemicals for
therapeutic use.Drugs usually work in one of four ways:
1. To replace or act as substitutes for missing chemicals
2. To increase or stimulate certain cellular activities
3. To depress or slow cellular activities
4. To interfere with the functioning of foreign cells, such as invading
microorganisms or neoplasms (drugs that act in this way are called
chemotherapeutic agents). Drugs can act in several different ways to
achieve these results.Receptor Sites
Many drugs are thought to act at specific areas on cell membranes called receptor
sites. The receptor sites react with certain chemicals to cause an effect within the
cell. In many situations, nearby enzymes break down the reacting chemicals and
open the receptor site for further stimulation. To better understand this process,
think of how a key works in a lock. The specific chemical (the key) approaches a
cell membrane and finds a perfect f it (the lock) at a receptor site. The interaction
between the chemical and the receptor site affects enzyme systems within the cell.
The activated enzyme systems then produce certain effects, such as increased or
decreased cellular activity, changes in cell membrane permeability, or alterations
in cellular metabolism. Some drugs interact directly with receptor sites to cause
the same activity that natural chemicals would cause at that site. These drugs are
called agonists. For example, insulin reacts with specific insulin-receptor sites to
change cell membrane permeability, thus promoting the movement of glucose into
the cellOther drugs act to prevent the breakdown of naturaal chemicals that are stimulating
the receptor site. Some drugs react with receptor sites to block normal stimulation,
producing no effect.Drug-Enzyme Interactions
Drugs also can cause their effects by interfering with the enzyme systems that act as
catalysts for various chemical reactions. Enzyme systems work in a cascade fashion,with one enzyme activating another, and then that enzyme activating another, until
a cellular reaction eventually occurs. If a single step in one of the many enzyme
systems is blocked, normal cell function is disrupted. Acetazolamide (Diamox) is a
diuretic that blocks the enzyme carbonic anhydrase, which subsequently causes
alterations in the hydrogen ion and water exchange system in the kidney, as well
as in the eye.Selective Toxicity
Ideally, all chemotherapeutic agents would act only on enzyme systems that are
essential for the life of a pathogen or neoplastic cell and would not affect healthy
cells. The ability of a drug to attack only those systems found in foreign cells is
known as selective toxicity. Penicillin, an antibiotic used to treat bacterial infections,
has selective toxicity. It affects an enzyme system unique to bacteria, causing
bacterial cell death without disrupting normal human cell functioning.Unfortunately, most other chemotherapeutic agents also destroy normal human
cells, causing many of the adverse effects associated with antipathogen and
antineoplastic chemotherapy. Cells that reproduce or are replaced rapidly (e.g.,
bone marrow cells, gastrointestinal [GI] cells, hair follicles) are more easily affected
by these agents. Consequently, the goal of many chemotherapeutic regimens is to
deliver a dose that will be toxic to the invading cells yet cause the least amount of
toxicity to the host.Self- assessment 2.10
1. There are four ways through which drugs usually work. Mention these 4
ways.2. The drugs administered to humans only affect the target cells, and never
harm the human cells because they were made in a specific way. TRUE
or FALSE2.11 Agonist drugs
Learning activity 2.11
Consult the library and read agonist drug, in pharmacology and to be able to
respond to the question of the scenario below.An associate nurse is caring an old woman with diabetes at home. After receiving
insulin injection, she asks associate nurse how the insulin will work to allow the
glucose to enter the cell.1. If you were the associate nurse in the scenario. What would you explain
to the woman?CONTENT SUMMARY
Drugs that interact directly with receptor sites to cause the same activity that natural
chemicals would cause at that site are agonists drugs.An agonist medication mimics the action of the signal by binding to and
activating a receptor.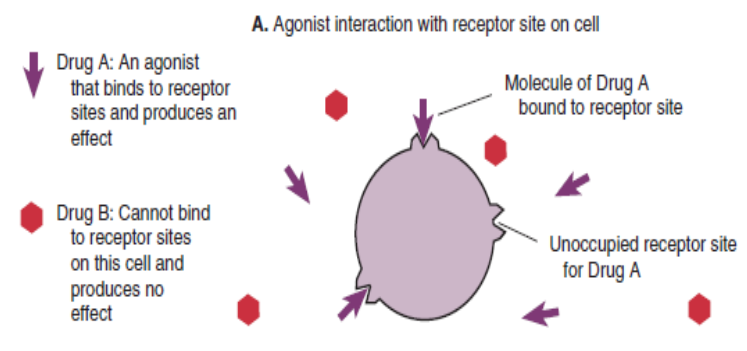
An agonist drug binds to receptor sites and produces an effect.
The insulin is an example of agonist drug as it reacts with specific insulin-receptor
sites to change cell membrane permeability, thus promoting the movement of
glucose into the cell. This is the same action as natural insulin would do in normal
human body.Full agonists are drugs when administered at concentrations sufficient to saturate
the receptor pool, can activate their receptor-effector systems to the maximum
extent of which the system is capable and this causes a shift of almost all of the
receptor pool. On the other hand, partial agonists. The term partial agonist or
agonist-antagonist drug describes a medication that produces a weaker, or less
efficacious, response than an agonist. It binds to the same receptors and activate
them in the same way but do not evoke as great a response, no matter how high
the concentration.Example of full agonist effect in clinical application is administration of bethanechol
(Urecholine). It binds to acetylcholine receptors in the autonomic nervous system
and produces the same actions as acetylcholine.Self- assessment 2.11
1. Define agonist drug
2. Differentiate full agonist from partial agonist drug2.12 Drug Antagonists
Learning activity 2.12
During your clinical placement in the hospital, you observe a nurse giving a drug
named atropine to the patient with very slow heart rate. Your colleague asks the
nurse how the atropine will increase the heart rate. The nurse explains in short
word that the atropine is antagonist of acetylcholine a neurotransmitter of the
parasympathetic nervous system that can slow the heart rate.1. Visit the library and read the content of antagonist drug and briefly
describe what the antagonist drug is.CONTENT SUMMARY
Antagonism is an interaction between two or more drugs that have opposite
effects on the body. Antagonist may block or reduce the effectiveness of one or
more of the drugs. An antagonist is a medication that typically binds to a receptor
without activating them, but instead, decreases the receptors ability to be activated
by other agonist. That drug will occupy a receptor and prevent the endogenous
chemical from acting. Antagonists often compete with agonists for the receptor
binding sitesA competitive antagonist is a drug that binds to the same receptor sites as another
drug and prevents it from binding.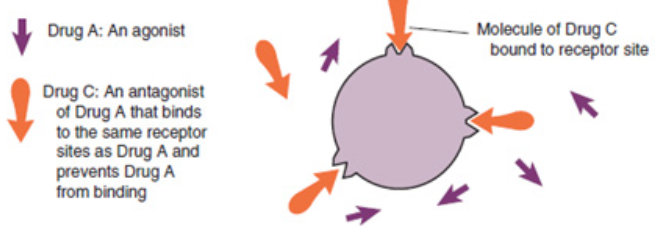
A noncompetitive antagonist is a drug that binds to different receptor sites from
another drug but still prevents that drug from binding.Not all antagonism is associated with receptors. Functional antagonists inhibit the
effects of an agonist not by competing for a receptor but by changing pharmacokinetic
factors. For example, antagonists may slow the absorption of a drug. By speeding
up metabolism or excretion, an antagonist may enhance the removal of a drug from
the body. An other example of antagonism include antidote effect on drugsThe relationships that occur between agonists and antagonists explain many of the
drug–drug and drug–food interactions that occur in the body.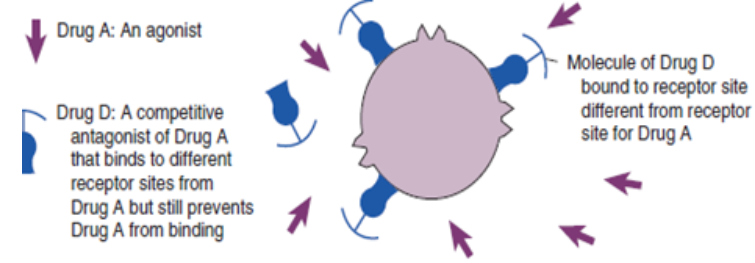
An example of antagonist effect in clinical application is the use of the drug
atropine which competes with acetylcholine for specific receptors associated with
the autonomic nervous system. If the dose is high enough, atropine will inhibit the
effects of acetylcholine, because acetylcholine cannot bind to its receptors.Self- assessment 2.12
1. Define a drug antagonist
2. Differentiate competitive from non- competitive antagonist drug2.13 Pharmacokinetics in special population
Learning activity 2.13
While you were in clinical practice in consultation room, you saw a senior nurse
played attention while prescribing the drugs to the children and old people than
other group of people between 20 to 50 years old.1. Visit the library, read the books of pharmacology on pharmacokinetics
special considerations and come up with a summary of why Children
often require different doses of drugs than adults.CONTENT SUMMARY
Pharmacokinetics are typically dependent on a variety of physiological variables
(e.g., age, ethnicity, or pregnancy) or pathological conditions (e.g., renal and hepatic
insufficiency, cardiac dysfunction, obesity, etc.).To providing safe and effective medications, pediatric drug therapy represent a
great challenge to the health professionals. Children often require different doses
of drugs than adults because children’s bodies often handle drugs very differently
from adults’ bodies. In some cases, a pediatric dose is suggested, but in many
cases it will need to be calculated based on the weight and the age of the child.Medications can affect the fetus either by interfering with some important function
in the mother which indirectly damages the fetus or by pass across the placenta or
acting directly on the fetus.Most drugs cross the placenta,30% of pregnant women take drugs and 10% take
drugs in the first trimester when the fetus is more vulnerable. It is important to
discover which drugs can produce fetal damage and which are safe to use but it is
difficult because in the period of implantation (5-15 days): Drug toxicity can result in
abortion, in Embryonic stage (15 to 55days): Embryo is changing from a group of
cells into a recognizable human being.The embryo is particularly susceptible to drug toxicity at this time and leads to fetal
malformation or teratogenesis (a process by which congenital malformations
are produced in an embryo or fetus). Fetogenic stage (55 to birth): Drug damage
is less likely but still possible, at Delivery: Drugs may interfere with labour and
modify the behaviour of neonates immediately after birth, Food drug administration
indicate the potential or actual teratogenic effects of a drug.The New-borns are unable to break down drugs as effectively older children or adults
do. Example the accumulation of chloramphenicol can cause grey syndrome due
to collapse of circulation.The period from birth to adolescent is characterised by dramatic changes in physical
growth, psychosocial development and sensitivity to drugs. Old persons are among
the most consumers of drugs. Yet their metabolism changes with age.Elder people have fewer albumins in the blood, with certain drug, less protein bound
and more are free in the blood and tissue fluids and can therefore produce a greater
pharmacological effect.With advanced age, liver enzyme decreases blood supply especially to liver
consequently the absorption decreases, as result some drugs may therefore be
more slowly broken down and their blood concentration may rise to toxic levels.Drugs are also excreted via the kidney. Old age, sometimes associated with kidney
diseases, leads to a decline in renal function, so that by the age of 80 years, renal
function is only half than at age 40. This again may cause drug accumulation in the
Body and evidence that certain systems become more sensitive to drug action with
advancing years.Self- assessment 2.13
1. What are the special considerations in case of pharmacokinetics during
drug administration?2. Why drug toxicity can rise in the people over 80 years old?
2.14 Pharmacodynamics in Special population
Learning activity 2.14
1. Visit the library and read the book of pharmacology on pharmacodynamics
special population and summarize how the medication can affect the
fetus.CONTENT SUMARY
All living organisms function by a series of complicated, continual chemical reactions.
When a new chemical enters the system, multiple changes in and interferences
with cell functioning may occur. To avoid such problems, drug development works
to provide the most effective and least toxic chemicals for therapeutic use.The reactions changes depending on many factors including receptors site age
and personal health status. Pregnant women, children and older persons are
special population for whom attention must be taken when administering them the
medication.The use of drugs in pregnancy is complicated by the potential for harmful effects
on the growing fetus, altered maternal physiology. Because experience with many
drugs in pregnancy is severely limited, it should be assumed that all drugs are
potentially harmful until sufficient data exist to indicate otherwise.Some drugs’ effect may be serious to the pregnant woman and may even be fatal
for the unborn baby. Again when administered during delivery drugs may change
neonates behaviors even lead to immediate complications. Medications can affect
the fetus either by interfering with some important function in the mother which
indirectly damages the fetus or by pass across the placenta or acting directly on
the fetus.There are drugs that have toxicity that when given during implantation period
they cause abortion. Other may cause fetal malformation or teratogenesis when
administered during the embryonic period of pregnancy. Drugs may interfere with
labor and modify the behavior of neonates immediately after birth.For every pregnant woman, it is imperative to avoid giving drugs as possible in
the first 3 months of pregnancy, give drugs at the lowest effective dose for as a
short time as possible, avoid recently introduced drugs if possible, be sure that
every female you attempt to give medication is pregnant or not, read drug
risk category before administration of any drug to a pregnant female.Most drugs pass into breast milk, but at a very low and innocuous concentration.
Generally, drugs should be avoided by nursing mothers, but if the drug is essential
the baby should feed before the mother take drugs, then when blood levels will be
low. Certain drugs should not be used by nursing mothers and, if unavoidable, will
require transfer to bottle feeding.The liver of children is immature and depending on age some are inactive this mark
the difference in drug metabolism in the body that finally may cause accumulation
and increase drug toxicity.Children have immature renal system and it is difficult to excrete drugs. This
increase risk for toxicity.Factors affecting pharmacodynamics of a drug in children are summarized
as below:
– Reduced gastric acidity some medication that require acid environment to be
broken are not well metabolized.– Small muscle mass: Drugs administered in intramuscular should be at lower
dose to allow absorption.– Thin stratum corneum: Topical application of medication can be easily
absorbed and when large amount is applied toxicity be present. Again special
attention when administered subcutaneous medication is taken as it is very
easy to reach the muscle.– High proportion of water in body: Water soluble drugs are highly absorbed
and lipid soluble drugs are poorly absorbed.– Reduced protein-binding capability this limit some drug absorption and
distribution to the whole body.– Unpredictable hepatic enzymes production
– Immature renal system
Self- assessment 2.14
1. What are the considerations a nurse might take to avoid harmful drug
effects on a pregnant woman?2.15 Dose-Response Relationships
Learning activity 2.14
An associate nurse was assigned to care for an old man on palliative care. The
man is receiving morphine as analgesic drug 5 mg Subcutaneous route (SC).
Today associated nurse visited the client and the client tells him that the dose
he is receiving do as nothing. Associated nurse called the physician, and the
physician order to increase the dose as 8mg. The client after receiving 8mg
dose, report the relief of pain.1. In your view, why 5 mg was not reducing pain and 8 mg reduce the pain?
CONTENT SUMMARY
How does a patient respond to varying doses of a drug? Common sense would
suggest that a larger dose would produce more drug effect.An antihypertensive drug would cause a greater reduction in blood pressure if the
dose was increased from 50 to 100 mg. These simple examples describe the dose–
response relationship, one of the most fundamental concepts in pharmacology.Examining and comparing dose–response curves can yield a large amount of
information about a drug. A dose–response curve plots the drug dose administered
to the patient versus the intensity or degree of response obtained.There are three distinct phases of a dose–response curve that indicate essential
pharmacodynamics principles.Phase 1 occurs at the lowest doses.
The flatness of this portion of the curve indicates that few target cells have been
affected by the drug; doses that are too small will not produce a therapeutic effect.Phase 2 is the rising, straight line portion of the curve. In this portion, there is a
linear relationship between the amount of drug administered and the degree of
response obtained from the patient. For example, if the dose is doubled, twice as
much response may be obtainedThis is the most desirable range of doses for pharmacotherapeutics, because
giving more drug results in proportionately more effect; a lower drug dose gives
less effect.In phase 3 increasing the drug dose produces no additional therapeutic response a
plateau has been reached. This may occur for a number of reasons. One possible
explanation is that all the target receptors for the drug are occupied. It could also
mean that the drug has brought 100% relief, such as when a migraine headache
has been terminated; giving higher doses produces no additional relief.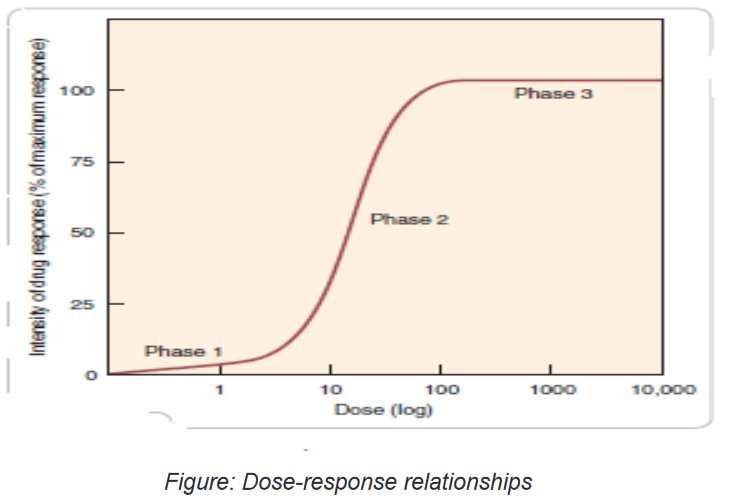
Self- assessment 2.15
1. After reading the content of the lesson, what do you understand by dose
response relationship?2.16 Potency of drug
Learning activity 2.16
1. Using library Pharmacology textbook/internet search the term Potency of
the drug and make short note on term Potency of drug.CONTENT SUMMARY
The concept of potency is first fundamental ways to compare medications
within therapeutic and pharmacologic classes. Pharmacologic potency can
largely determine the administered dose of the chosen drug. For therapeutic
purposes, the potency of a drug should be stated in dosage units, usually in terms of
a particular therapeutic end point may be used in comparing one drug with another.Potency is an index of how much drug must be administered to elicit a desired
response. A drug that is more potent will produce its therapeutic effect at a lower
dose, compared to another drug in the same class.Thus, potency is a way to compare the doses of two independently
administered drugs in terms of how much is needed to produce a particular
response.If a drug were of extremely low potency, we might need to administer that drug
in huge doses multiple times a day to achieve beneficial effects. In this case, an
alternative drug with higher potency would be desirable. Fortunately, it is rare for
a drug to be so lacking in potency that doses of inconvenient magnitude need be
given. The only consequence of having greater potency is that a drug with greater
potency can be given in smaller doses.Which is more important to the outcomes of pharmacotherapy: potency or efficacy?
Perhaps the best way to understand these important concepts is to use the specific
example of headache pain. Two common analgesic therapies are ibuprofen 200mg,
and aspirin 650mg. The fact that ibuprofen relieves pain at a lower dose indicates
that it is more potent than aspirin.In clinical practice the term potency is often misused to indicate a more
effective drug. The nurse should remember the correct definitions of the words
potency and efficacy and try to incorporate them into clinical practice.In everyday, people tend to use the word potent to express the pharmacologic
concept of effectiveness. That is, when most people say, “This drug is very potent,”
what they mean is, “This drug produces powerful effects.” They do not mean, “This
drug produces its effects at low doses.” In pharmacology, we use the words potent
and potency with the specific and appropriate terminology.Self- assessment 2.16
1. What does “DRUG POTENCY” mean?
A. A measure of how tightly a drug bind to plasma proteins
B. A measure of how tightly a drug bind to a receptor
C. A measure of inhibiting potency of a drug
D. An index of how much drug must be administered to elicit a desired
response.2.17 Efficacy of drug
Learning activity 2.17
1. Using library Pharmacology textbook/internet search the term Efficacy of
the drug and make short note on term Efficacy of drug.CONTENT SUMMARY
The concept of Efficacy is a second fundamental ways to compare medications
within therapeutic and pharmacologic classes, which is defined as the greatest
maximal response that can be produced from a particular drug or defined as the
largest effect that a drug can produce. Maximal efficacy is indicated by the height
of the dose-response curve. The maximal efficacy of a drug is obviously crucial for
making clinical decisions when a large response is needed. It may be determined
by the drug’s mode of interactions with receptors (as with partial agonists,
described above) or by characteristics of the receptor-effector system involved.
Thus, therapeutic efficacy may be affected by the characteristics of a particular
drug-receptor interaction, but it also depends on a host of other factors.The best way to understand this important concept is to use the specific example
of headache pain. Two common analgesic therapies are ibuprofen 200mg, and
aspirin 650mg. The fact that ibuprofen relieves pain at a lower dose indicates that it
is more potent than aspirin. At the given doses, however, both are equally effective
at relieving headaches; thus they have the same sufficient efficacy to bring relief.
Morphine has a greater efficacy than aspirin or ibuprofen and could effectively treat
this type of pain. From a pharmacotherapeutic perspective, efficacy is almost
always more important than potency. In the preceding example, the average
dose is unimportant to the patient, but headache relief is essential.As another comparison, the patient with cancer is much more concerned
with how many cancer cells have been killed (efficacy) than with the dose the
nurse administered (potency).
Within a pharmacologic class, not all drugs are equally effective at treating a disorder.
For example, some antineoplastic drugs kill more cancer cells than others; some
antihypertensive agents lower blood pressure to a greater extent than others; and
some analgesics are more effective at relieving severe pain than others in the same
class. Furthermore, drugs in the same class are effective at different doses: one
antibiotic may be effective at a dose of 1mg/kg, whereas another is most effective
at 100mg/kg.A drug with very high maximal efficacy is not always more desirable than a drug
with lower efficacy. Recall that we want to match the intensity of the response to
the patient’s needs. It is important to note that the potency of a drug implies nothing
about its maximal efficacy! Potency and efficacy are completely independent
qualities.Drug A can be more effective than drug B even though drug B may be more potent.
Also, drugs A and B can be equally effective even though one may be more potent.
The only consequence of having greater potency is that a drug with greater potency
can be given in smaller doses.In deciding which of two drugs to administer to a patient, the prescriber must
usually consider their relative effectiveness rather than their relative potency. It is
important to distinguish between a drug’s potency and its efficacy for clinical use. To
choose among drugs and to determine appropriate doses of a drug, the prescriber
must know the relative pharmacologic potency and maximal efficacy of the drugs
in relation to the desired therapeutic effect. The clinical effectiveness of a drug
depends not on its potency (EC50), but on its maximal efficacy and its ability to
reach the relevant receptors. This ability can depend on its route of administration,
absorption, distribution through the body, and clearance from the blood or site of
action.Self- assessment 2.17
1. The term “drug efficacy” means:
A. Two drugs combine with one another to form an inactive compound
B. Two drugs combine with one another to form a more active compound
C. The greatest maximal response that can be produced from a particular
drug or defined as the largest effect that a drug can produce
D. Two drugs combine with one another to form a more water-soluble
compound.2.18 Therapeutic index
Learning activity 2.18
1. Read the book of pharmacology, discuss on therapeutic index (using
library books) and make short notes.CONTENT SUMMARY
The therapeutic index (TI; also referred to as therapeutic ratio) is a quantitative
measurement of the relative safety of a drug. It is a comparison of the amount of
a therapeutic agent that causes the therapeutic effect to the amount that causes
toxicity. The related terms therapeutic window or safety window refer to a range
of doses which optimize between efficacy and toxicity, achieving the greatest
therapeutic benefit without resulting in unacceptable side-effects or toxicity.The larger the therapeutic index (TI), the safer the drug is. If the TI is small (the
difference between the two concentrations is very small), the drug must be dosed
carefully and the person receiving the drug should be monitored closely for any
signs of drug toxicity.In the early days of pharmaceutical toxicology, TI was frequently determined in
animals as lethal dose of a drug for 50% of the population (LD50) divided by the
minimum effective dose for 50% of the population (ED50).Self- assessment 2.18
1. What do you understand by therapeutic index?
2. What the nurse have to do before and after administrating the drug which
have small therapeutic index?
3. The term therapeutic window or safety window refer to a range of doses
which optimize between:
A. Efficacy and toxicity
B. Efficacy and Lethal
C. Loading and maintenance
D. Potency and toxicity2.19 Inter patient Variability
Learning activity 2.19
1. Read the book of pharmacology (using library books), discuss on
therapeutic index Inter patient Variability and make note.CONTENT SUMMARY
Once drugs are administered, certain patients fail to react to treatment even when
systemic exposure to the drug is within the range associated with therapeutic
response. The main reasons are many but most of the time misdiagnosis of
the disease or individual lacking the therapeutic target or inability to express a
satisfactory response.The reasons behind patient difference in responsiveness to a given dose of a
drug are many. They include genetics, disease, age, gender, body weight, drugs
given concomitantly, and various behavioral and environmental factors. Age, body
weight, disease, and concomitantly administered drugs are important because they
are measurable sources of variability that can be taken into account. Gender-
linked differences in hormonal balance, body composition, and activity of certain
enzymes manifest themselves in differences in both pharmacokinetics and
responsiveness, but overall, the effect of gender is small. Although inheritance
accounts for a substantial part of the differences in response among individuals
for many drugs, much of this variability is still largely unpredictable, particularly in
regard to pharmacodynamics.The examples of variability in drug response so far have been of the therapeutic
effect of the drug, but the situation equally applies to adverse effects. For some
relatively minor adverse effects, variability may be as great as, or even greater
than, that for the therapeutic effect, particularly when they are associated with the
inherent pharmacologic property of the drug (side effects), such as dryness of mouth
experienced with some sympathomimetic nasal decongestants. Frequent side
effects are also invariably experienced by patients undergoing chemotherapy during
cancer treatment. However, in many other therapeutic settings, moderate to severe
side effects are much less frequently experienced. Occasionally, the frequency of
an adverse effect is so low that it is only detected with any significance when tens
of thousands, if not millions, of patients have been treated with the drug. Even so,
there is still some relationship between the likelihood and severity of an adverse
effect and the exposure to the drug, although establishing it with any confidence
may be difficult. The degree and relative contribution of pharmacokinetics and
pharmacodynamics to variability in response within a patient population vary with
the drugSelf- assessment 2.19
1. hat are the reasons behind patient difference in responsiveness to a
given dose of a drug?
2. Explain how gender can affect drug response?2.20 End unit assessment
End Unit assessment 2
1. Which of the following statements is true with regard to the meaning of
selective toxicity of an antibiotic?
A. The ability of the anti-infectious agent to affect both microbial and host
cells at the same time
B. The ability of anti-infectious agent to affect the host cell with few effects
to the microbial cell
C. The ability of an anti-infectious agent to affect the bacterial cell wall
since the human cell does also have the cell wall
D. The ability of the anti-infectious agent to affect the infectious agent’s cell
without affecting the host (human) cell2. Which of the following pharmacological terms deals with Absorption,
distribution, Metabolism and Elimination (Excretion) of drugs?
A. Pharmacodynamics
B. Pharmacognosy
C. Pharmacokinetics
D. Pharmacopoeia3. Which of the following assertions describes a teratogenic drug?
A. The drug that can produce severe adverse reactions
B. The drug that can impact negatively the elderly
C. The drug that can cause congenital malformation
D. That drug that have a broad spectrum of activity4. In pharmacology, “drug tolerance” means:
A. A potential maximum therapeutic response which a drug can produce if
used at right dose
B. A decreased response to a drug, requiring an increase in dosage to
achieve the desired effect
C. An increased response to a drug, requiring an increase in dosage to
achieve the desired effect
D. A margin between the therapeutic dose and lethal dose of any given
antibiotic medication5. All of the following statements about efficacy and potency are true
EXCEPT:
A. Efficacy is usually a more important clinical consideration than potency
B. Efficacy is the maximum effect of a drug
C. Potency is a comparative measure, refers to the different doses of two
drugs that are needed to produce the same effect
D. The ED50 is a measure of drug’s efficacy6. What does “pharmacokinetics” include?
A. Complications of drug therapy
B. Drug biotransformation in the organism
C. Influence of drugs on metabolism processes
D. Influence of drugs on genes7. Pharmacodynamics involves the study of the following?
A. Mechanisms of drug action
B. Biotransformation of drugs in the organism
C. Distribution of drugs in the organism
D. Excretion of drug from the organism8. If an agonist can produce submaximal effects and has moderate efficacy
it’s called:
A. Partial agonist
B. Antagonist
C. Agonist-antagonist
D. Full agonist